一种[1,4,5]氧二氮杂庚烷的制备方法及应用与流程
- 国知局
- 2024-06-20 11:12:45
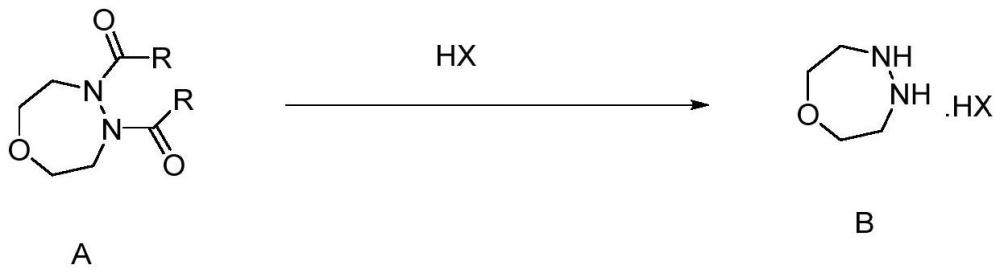
本发明涉及一种[1,4,5]氧二氮杂庚烷的制备方法及应用,属于农药化学与化工。
背景技术:
1、[1,4,5]氧二氮杂庚烷,cas:746595-79-9,别名[1,4,5]氧二氮杂卓,是一种重要的合成中间体,特别是在农药的合成的领域。例如[1,4,5]氧二氮杂庚烷是合成除草剂唑啉草酯的重要中间体(wo9947525)。
2、wo9947525(cn1185234c)公开了一种由n,n'-二叔丁氧羰基-[1,4,5]-氧二氮杂卓与氢溴酸在乙醚中反应来制备[1,4,5]氧二氮杂庚烷氢溴酸盐的工艺,反应温度20℃搅拌22小时,反应温度35℃搅拌27小时。虽然该工艺反应温度低,但是所用的保护基团原料boc2o价格昂贵,分子量大、脱保护时产生大量的废料,使用的溶剂乙醚闪点低、存在安全隐患。不仅如此,该工艺反应总时间长(48小时)、效率较低。
3、wo03051853a(cn1279032c)公开了以含有4,5-二酰基-[1,4,5]-氧二氮杂卓结构的有机物为原料,采用氢卤酸优选地是氯化氢和溴化氢,在极性溶剂中,在升高的温度下(43-50℃),反应脱去酰基生成含有[1,4,5]-氧二氮杂卓结构的有机化合物。其中所述的极性溶剂优选地使用沸点在100℃以上的醇,例如正丁醇、正戊醇、环己醇、苯酚、苯甲醇,乙二醇,二甘醇,甘油,甲氧基异丙醇和乙氧基乙醇的工艺。该工艺虽然解决了上述boc2o价格昂贵,分子量大、脱保护时废料多的问题,但是依然存在较多问题,如反应周期依然太长(18-22小时),溶剂的沸点要求高(沸点100℃以上),产物中有溶剂包夹导致产物纯度不高(纯度约90%,含溶剂约9%)等问题。
4、wo2006045587(cn101039926b)公开了由含有4,5-二酰基-[1,4,5]-氧二氮杂卓结构的有机物为原料,与碱金属或碱土金属的氢氧化物、碳酸盐和醇化物的碱,在选自水、具有高于100℃的沸点的醇、dmso、环丁砜、nmp、dma或dmf或其混合物的极性溶剂中,反应脱去酰基生成含有[1,4,5]-氧二氮杂卓结构的有机化合物。其中所述的反应可以在有可溶于反应混合物的盐存在下进行的工艺。该工艺反应温度要求较高,且只有在以水为溶剂时才能取得较高的收率(65-90%),同时需要使用大量的有机盐,易产生大量废固。除此以外由于产物[1,4,5]氧二氮杂卓在水中溶解性极好,导致产物分离困难,需要经过多次萃取分离,既增加了生产成本,又产生大量废液。
5、cn108264492b和cn108264493b公开了以含有4,5-二酰基-[1,4,5]-氧二氮杂卓结构的有机物为原料,采用氢氧化钾为碱,在非极性溶剂(甲苯或二甲苯)中,在100-130℃的温度下,反应脱去酰基生成含有[1,4,5]-氧二氮杂卓。该工艺报导的收率高,报导的反应时间较短,但反应温度高,超过100℃,实施例温度都为甲苯回流温度130℃,我们发现该反应在降低温度下不反应,只有温度达到较高的反应温度如130℃时,会快速反应,并集中放热,这将给工业放大带来很大的安全风险,存在脱保护时废料多的问题,特别是会产生大量的含甲苯或二甲苯的氢氧化钾和乙酸钾的混合废盐,该废盐成分复杂,需做危废处理,成本高;同时非极性溶剂(甲苯或二甲苯)沸点高,也会给后续反应步骤中除溶剂带来能耗较高的问题。
6、in201821039267a公开了以4,5-二乙酰基-[1,4,5]-氧二氮杂卓为原料,采用氯化氢,在极性溶剂中,在升高的温度下(45-60℃),反应脱去乙酰基生成含有[1,4,5]-氧二氮杂卓的方法。反应周期依然太长(47小时),产物收率不高(约38-47%)。
7、因此,需要开发呈现一种或多种改进的[1,4,5]氧二氮杂庚烷的制备方法。
技术实现思路
1、本发明的目的是为解决现有技术中[1,4,5]氧二氮杂庚烷的制备方法存在的问题。
2、为达到解决上述问题的目的,本发明所采取的技术方案是提供一种[1,4,5]氧二氮杂庚烷的制备方法,包括以下步骤:将式(a)所示的4,5-二酰基-[1,4,5]-氧二氮杂卓结构的有机物溶于有机溶剂中,加入环状冠醚类催化剂或季铵盐催化剂,通入氢卤酸(hx)进行反应,反应结束后,经后处理制备得到式(b)所示的[1,4,5]氧二氮杂庚烷的氢卤酸盐。
3、
4、优选地,所述的有机溶剂为具有低于100℃的沸点的极性溶剂;所述的极性溶剂进一步优选为醇,更优选为叔丁醇,正丙醇,异丙醇,乙醇和甲醇中的至少一种,有机溶剂可回收套用。
5、优选地,所述的式(a)化合物与有机溶剂的质量比为1:1.0-9.0,进一步优选为1:3.0-7.0。
6、优选地,所述的环状冠醚类催化剂为18冠6,季铵盐催化剂为四丁基硫酸氢铵。
7、优选地,所述的式(a)化合物与所述的催化剂的质量比为1:0.005-0.3,进一步优选为1:0.01-0.2。
8、优选地,所述的式(a)化合物与氢卤酸的摩尔比为1:2.0-9.0,进一步优选为1:3.0-8.0。
9、优选地,所述的反应温度为35~80℃;进一步优选为40~60℃。
10、优选地,所述的反应总时间为2~12小时;进一步优选为4~8小时。
11、优选地,所述的后处理步骤包括通过降温使产物从反应体系中沉淀析出,并通过在氮气保护的过滤器中过滤后得到产物。
12、本发明也通过实验验证了该制备方法得到的[1,4,5]氧二氮杂庚烷的氢卤酸盐,经过进一步的反应,可转化为除草剂如唑啉草酯。
13、相比现有技术,本发明具有如下有益效果:
14、1、本发明的制备方法,在较为温和的反应温度下,反应速度明显大幅提升,反应时间明显缩短,同时反应的转化率好,收率较高,收率最高可达96.4%。
15、2、本发明的制备方法中,采用的沸点相对较低的醇类溶剂,这类溶剂易回收,同时回收能耗较低,产品中溶剂残留明显变小,产品纯度和质量含量明显提高,同时改善了现有技术提及的粘性晶体的搅拌效果不好的问题。
16、3、本发明的的制备方法中,采用的催化剂都是常用的价格相对便宜的催化剂,且催化剂用量小,对该反应的催化效果好,显著的提高了反应速度,同时降低了反应杂质的产生,提高了产品品质。同时明显改善了现有技术提及的[1,4,5]-氧二氮杂卓的氢卤酸盐的稳定性不太好,不便保存的问题。
17、4、本发明所用的后处理工艺简单,高效,晶体粘性变小,过滤效果好,改善了现有技术提及的[1,4,5]-氧二氮杂卓的氢卤酸盐的分离效果不太好的问题,同时密闭保存稳定性好,母液可回收套用,固废少,成本节约,经济性好。
18、综上所述,本发明的方法与现有[1,4,5]氧二氮杂庚烷的生产方法相比,反应效率高,反应时间短,得到的产物的收率高,纯度高,含量高,工艺简单,固废少,成本节约,经济性好,更适合大规模的工业化生产。
技术特征:1.一种[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,包括以下步骤:将4,5-二酰基-[1,4,5]-氧二氮杂卓类有机物溶于有机溶剂中,加入环状冠醚类催化剂或季铵盐催化剂,通入氢卤酸进行反应,反应结束后,经后处理制备得到[1,4,5]氧二氮杂庚烷的氢卤酸盐。
2.如权利要求1所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的有机溶剂为具有低于100℃的沸点的极性溶剂。
3.如权利要求2所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的极性溶剂为醇。
4.如权利要求3所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的醇为叔丁醇,正丙醇,异丙醇,乙醇和甲醇中的至少一种。
5.如权利要求1所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的环状冠醚类催化剂为18冠6,季铵盐催化剂为四丁基硫酸氢铵。
6.如权利要求1所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的4,5-二酰基-[1,4,5]-氧二氮杂卓类有机物与所述的催化剂的质量比为1:0.005-0.3。
7.如权利要求1所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的4,5-二酰基-[1,4,5]-氧二氮杂卓类有机物与氢卤酸的摩尔比为1:2.0-9.0。
8.如权利要求1所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的4,5-二酰基-[1,4,5]-氧二氮杂卓类有机物为4,5-二乙酰基-[1,4,5]-氧二氮杂卓或4,5-二丙酰基-[1,4,5]-氧二氮杂卓;所述的氢卤酸为氯化氢或溴化氢。
9.如权利要求1所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的反应温度为35~80℃;所述的反应总时间为2~12小时。
10.如权利要求1所述的[1,4,5]氧二氮杂庚烷的制备方法,其特征在于,所述的后处理步骤包括通过降温使产物从反应体系中沉淀析出,并通过在氮气保护的过滤器中过滤后得到产物。
技术总结本发明提供了一种[1,4,5]氧二氮杂庚烷的制备方法及应用,包括以下步骤:将4,5‑二酰基‑[1,4,5]‑氧二氮杂卓类有机物溶于有机溶剂中,加入环状冠醚类催化剂或季铵盐催化剂,通入氢卤酸进行反应,反应结束后,经后处理制备得到[1,4,5]氧二氮杂庚烷的氢卤酸盐。属于农药化学与化工技术领域。综上所述,本发明的方法与现有[1,4,5]氧二氮杂庚烷的生产方法相比,反应效率高,反应时间短,得到的产物的收率高,纯度高,含量高,工艺简单,固废少,成本节约,经济性好,更适合大规模的工业化生产。技术研发人员:王攀登,吴海琴,吴克崇,贾俊超,郭凯杰,王佳受保护的技术使用者:上海农帆生物科技有限公司技术研发日:技术公布日:2024/6/18本文地址:https://www.jishuxx.com/zhuanli/20240619/1219.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表