一种二膦钯配合物及其应用的制作方法
- 国知局
- 2024-06-20 11:27:09
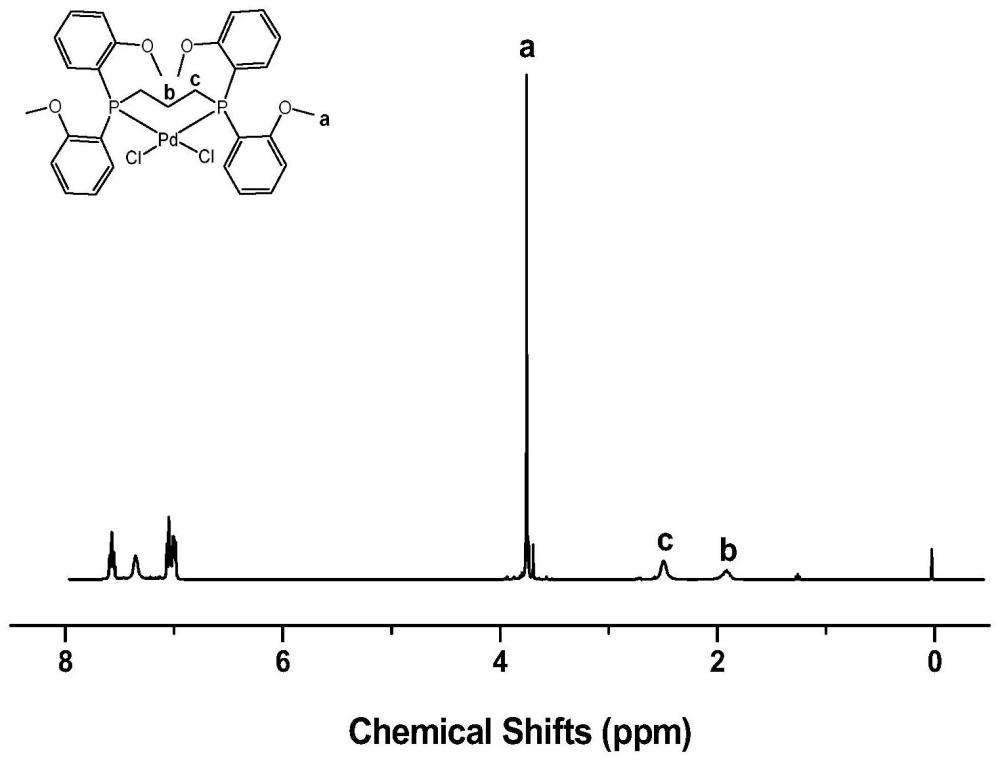
本发明涉及聚酮制备,具体涉及一种二膦钯配合物及其应用。
背景技术:
1、废旧塑料拥有稳定的物理化学结构,塑料制品在自然环境中可能数百年都不会分解,被称为“永久性垃圾”。现多采用填埋或焚烧两种方式处理废旧塑料:(1)填埋处理:不仅会占用大量土地,而且被占用土地长期得不到恢复,影响土地的可持续利用;(2)焚烧处理:废旧塑料燃烧时会产生二恶英或其他有毒气体,例如,聚氯乙烯燃烧时产生氯化氢,聚苯乙烯燃烧时产生甲苯。
2、光降解塑料是指通过光的作用可实现降解的塑料,该类塑料中的聚合物分子链在紫外线等光线照射下可激发电子活性,进而发生光化学反应,再加上大气环境中o2的影响,最终可发生光氧降解。在光化学作用下,光降解塑料的高分子链因遭到破坏而失去强度,材料发生脆化,并在风、雨等自然环境的作用下进一步细脆化,最终分解成为粉末融入土壤进入新一轮的生物循环。
3、光降解塑料技术已经较为成熟,每年的产量大幅度提升,其普遍应用于购物袋、垃圾袋、包装袋、塑料瓶以及农用地膜等,之后随着技术的不断成熟,也开始应用于新型快餐盒。光降解塑料的制备方法主要有两种。一种是在聚合物材料中融入光敏感物质,或者混入分解剂,常用的有金属化合物(金属盐、氧化物)、羰基化合物(二苯甲酮、对苯醌)等,多核芳香化合物(菲、六氢芘)也比较常用,这些添加剂在吸收光能后可产生自由基,也可以将激发态能量传递给聚合物,聚合物在激发态能量作用下产生自由基,最后促使聚合物在氧化作用下劣化,进而达到降解目的,称为添加性光降解材料。
4、另外一种是通过共聚的方法在聚合物分子链中加入光敏感基团,这样聚合物就可以具备光降解特性称为共聚合成型光降解材料。目前已经实现工业化生产的光降解塑料有乙烯酮共聚物、乙烯/co共聚物、接枝共聚物等,其中烯烃和co的共聚物较多。利用齐聚方式,将适当的光敏感基(如羧基、双键等)导入高分子结构内赋予材料光降解的特性。这种材料本身有光敏性能,光降解均匀、彻底。但其合成研究复杂,是研究的主要方向,以乙烯和一氧化碳的共聚物eco(聚酮)为例,其光降解按norrish i型及ii型反应进行。
5、n-i型
6、n-i1型
7、其中norrish i(n-i)型为主反应,被激发的羰基很容易与γ位上的氢形成六元环过渡态,因此,eco中羰碳基作为生色团,吸收光能并转变为化学能,发生光降解,造成分子量下降,强度减低,从而达到降解的目。
8、聚酮材料主要由乙烯和空气污染物co共聚而成,是一种非常环保的工程塑料。同时聚酮树脂的可降解性,能够有效解决塑料垃圾的污染问题,已成为可降解塑料研发热点之一。石油具有丰富的乙烯和co原材料,开发可降解聚酮材料具有显著的资源优势,同时也为中石油提供了一种新型的高附加值产品,具有显著的经济效益。乙烯和极性单体co共聚合成的可降解聚酮树脂,具有高冲击性能(与原有工程塑料尼龙、pbt相比耐冲击性强2.3倍以上),高耐磨损性(现存的最坚固材料聚缩醛相比耐磨损性高14倍以上),高阻隔性(与被使用为食品包装材料的evoh是同等水准),高阻燃性(与尼龙相比,可节省阻燃剂含量50%),以及杰出的抗化学性和耐热性;同时主链上存在的羰基赋予其优良的光降解性。因此,聚酮材料广泛应用于轴承、齿轮、阻隔管、传送带皮带、水栓配件等特种工程塑料领域;也可用于汽车及电子电器元件领域,如汽车燃油系统和引擎盖下罩、汽车外部组件、电子中的连接器、断路开关等。
9、cn114276669a公开了一种聚酮树脂复合材料及其制备方法,该聚酮树脂复合材料由包括以下质量份的组分制得:60-90份聚酮,10-40份abs树脂,5-10份季戊四醇硬脂酸酯,2-10份抗氧剂,0.5-1份光稳定剂。该专利将聚酮、abs树脂、季戊四醇硬脂酸酯、抗氧剂和光稳定剂以一定比例搭配制得聚酮树脂复合材料,能够拓宽加工区间、降低熔融加工温度、减少固化交联现象,从而改善其加工性能,同时保证其优异的物理机械性能。cn114410057a公开了一种能够降低阻燃剂使用量的abs阻燃合金及其制备方法,包括一种能够降低阻燃剂使用量的abs阻燃合金,包括以下原料组成:abs树脂:30%-40%;聚酮树脂:10%-45%;聚乙烯蜡:5%-10%;硬脂酸钙:5%-10%;增韧剂5%-20%;溴系阻燃剂10%-15%;阻燃协效剂3%-6%;苯乙烯-丙烯腈-甲基丙烯酸缩水甘油酯共聚物2%-5%;抗氧剂:5%-10%;白油:2%-10%,其涉及阻燃高分子合金材料技术领域。该专利能够降低阻燃剂使用量的abs阻燃合金及其制备方法,通过高速搅拌得到均匀混合的物料,便于后续处理的进行,能够降产出薄膜的颗粒状杂质,对颗粒进行挤出,便于得到能够降低阻燃剂使用量的合金,使得设备内部的残留物不易在凝固后出现堵塞的问题,对产出成品进行检测。但是,上述专利技术均存在一定的缺陷或不足之处:聚酮树脂复合材料的制备工程中co和金属中心很容易发生配位,得到高度稳定的co配合物,造成共聚物的活性显著降低,未提高共聚物的活性。
10、可见,在聚酮制备中,co和金属中心很容易发生配位,得到高度稳定的co配合物,造成共聚的活性显著降低。此外,co与乙烯在常规聚合中易形成完全交替共聚物,此类聚合物结构规整,结晶度高,在实际加工过程中加工难度大。
技术实现思路
1、为解决上述技术问题,本发明的目的在于提供一种具有边臂效应的二膦钯配合物,在配体中引入磷原子和氧原子,通过边臂功能基团与钯金属中心的协同作用,解决co与中心金属配位作用导致的聚合体系催化剂活性弱、产品分子量低等难题,增强乙烯与极性单体的共聚活性,提高催化剂活性,制备高分子量聚酮产品。
2、为达到上述目的,本发明提供一种二膦钯配合物,其化学结构式为:
3、
4、上述二膦钯配合物,优选地,所述二膦钯配合物的制备方法包括如下步骤:
5、s1:在氮气保护下,使亚磷酸二乙酯与2-甲氧基苯基溴化镁溶液(格氏试剂)在四氢呋喃中充分混合,然后进行第一反应,终止反应后,得到双-(2-甲氧基苯基)氧化膦;
6、s2:在氮气保护下,使所述双-(2-甲氧基苯基)氧化膦与1,3-二氯丙烷、氢氧化钾溶液、四丁基碘化铵在苯溶剂中进行第二反应,淬灭后得到1,3-双(二(2-甲氧基苯基)氧化膦)丙烷;
7、s3:在氮气保护下,使所述1,3-双(二(2-甲氧基苯基)氧化膦)丙烷与三氯硅烷、三丁胺、乙腈进行第三反应,淬灭后得到双膦配体;
8、s4:在氮气保护下,使所述双膦配体与氯化钯在二氯甲烷中进行第四反应,反应结束后加入析出剂,析出的固体即为所述二膦钯配合物。
9、二膦钯配合物二膦钯配合物上述二膦钯配合物的制备方法中,优选地,s1中,以亚磷酸二乙酯的用量为3-5ml为基准,2-甲氧基苯基溴化镁溶液的以溶质计的用量为100-180mmol,即2-甲氧基苯基溴化镁溶液中2-甲氧基苯基溴化镁纯物质的用量100-180mmol。
10、上述二膦钯配合物的制备方法中,优选地,s1中,2-甲氧基苯基溴化镁溶液的浓度为2-3mol/l。2-甲氧基苯基溴化镁溶液是一种格氏试剂,其中,2-甲氧基苯基溴化镁的结构式为溶剂为四氢呋喃。
11、上述二膦钯配合物的制备方法中,优选地,s1中,亚磷酸二乙酯与2-甲氧基苯基溴化镁溶液在0-10℃温度下混合。
12、上述二膦钯配合物的制备方法中,优选地,s1中,所述第一反应的温度为20-30℃,反应时间为2-3h。
13、上述二膦钯配合物的制备方法中,优选地,s1中,终止第一反应的方式为:将反应体系冷却至1-10℃,然后向其中依次滴加盐酸和甲基叔丁基醚并搅拌均匀。
14、上述二膦钯配合物的制备方法中,优选地,s1中,所述盐酸的浓度为0.1-0.2mol/l;以亚磷酸二乙酯的用量为3-5ml为基准,所述盐酸的用量为70-80ml,甲基叔丁基醚的用量为7-80ml。
15、上述二膦钯配合物的制备方法中,优选地,s1还包括从终止后的反应液中提取目标产物的步骤,包括:取有机相并去除其中水、有机溶剂和甲基叔丁基醚,得到油状物再用乙醚重结晶纯化,得到所述双-(2-甲氧基苯基)氧化膦。
16、上述二膦钯配合物的制备方法中,优选地,s2中,以双-(2-甲氧基苯基)氧化膦的用量为500-600mg为基准,所述1,3-二氯丙烷、以纯物质计的氢氧化钾、四丁基碘化铵的用量分别为0.1-0.2ml、0.3-0.5g和70-80mg。
17、上述二膦钯配合物的制备方法中,优选地,s2中,所述第二反应的温度为80-90℃,反应时间7-8h。
18、上述二膦钯配合物的制备方法中,优选地,s2中,淬灭所述第二发应的方式为:将反应体系冷却至20-30℃,然后向其中加入水,完成淬灭。
19、上述二膦钯配合物的制备方法中,优选地,s2还包括从淬灭后的反应液中提取目标产物的步骤:用有机溶剂萃取反应液,除去其中水和有机溶剂,所得产物过柱分离纯化,得到所述1,3-双(二(2-甲氧基苯基)氧化膦)丙烷。
20、上述二膦钯配合物的制备方法中,优选地,s3中,以所述1,3-双(二(2-甲氧基苯基)氧化膦)丙烷的用量为300-400mg为基准,所述三氯硅烷、三丁胺、乙腈的用量分别为0.5-1ml、1-2ml和7-8ml。
21、上述二膦钯配合物的制备方法中,优选地,s3中,所述第三反应的温度为70-80℃,反应时间为5-6h。
22、上述二膦钯配合物的制备方法中,优选地,s3中,淬灭所述第三反应的方式为:将反应体系冷却至20-30℃,向其中加入氢氧化钠溶液,完成淬灭。
23、上述二膦钯配合物的制备方法中,优选地,s3中,所述氢氧化钠溶液的质量浓度为20-30%;以所述1,3-双(二(2-甲氧基苯基)氧化膦)丙烷的用量为300-400mg为基准,所述氢氧化钠溶液的用量为2-3ml。
24、上述二膦钯配合物的制备方法中,优选地,s3还包括从淬灭后的反应液中提取目标产物的步骤:用有机溶剂萃取反应液,除去其中水和有机溶剂,所得产物过柱分离纯化,得到所述双膦配体。
25、上述二膦钯配合物的制备方法中,优选地,s4中,所述双膦配体与氯化钯的摩尔比为1-3:1-3优选为1:1。
26、上述二膦钯配合物的制备方法中,优选地,s4中,所述第四反应的温度为20-30℃,反应时间1-2h。
27、上述二膦钯配合物的制备方法中,优选地,s4中,所述析出剂为正戊烷与乙醚的混合物,正戊烷与乙醚的体积比为1-2:1-2。
28、上述二膦钯配合物的制备方法中,优选地,在s1-s4各步骤的反应之后,各步骤的有机相均经过干燥处理,处理方式为采用无水硫酸钠干燥除水。
29、上述二膦钯配合物的制备方法中,优选地,所述二膦钯配合物的结构式为
30、
31、上述二膦钯配合物的制备方法中,优选地,具体包括如下步骤:其中,所有操作严格按照无水无氧操作进行:
32、(1)将250ml支口烧瓶真空干燥2-3小时,充入氮气密封;向其注入50-60ml浓度为2-3mol/l的2-甲氧基苯基溴化镁溶液(格氏试剂),接着将溶液在冷冻槽中冷却至0℃;将3-5ml的亚磷酸二乙酯、四氢呋喃(thf)溶剂10-20ml通过滴液漏斗缓慢滴加加入格氏试剂的烧瓶,滴加完毕后溶液在0℃下搅拌30-60min,然后从冷冻槽中取出自然升温到室温,继续在常温下搅拌2-3小时后,再次放入冷冻槽中冷却到0℃然后滴加入70-80ml浓度为0.1-0.2mol/l的盐酸,继续加7-80ml甲基叔丁基醚搅拌5-10min终止反应产物通过倾析分离出上部有机相,下面的含固体的水相进一步通过100ml的ch2cl2萃取分离有机相,与上层有机相合并有机相加入无水硫酸钠干燥除水,过滤,旋转蒸发除掉有机溶剂,继续在真空70度下除掉甲基叔丁基醚,得到淡黄色油状物,用乙醚重结晶纯化,得到白色固体,即双-(2-甲氧基苯基)氧化膦。
33、该步骤的反应式如式(1)。
34、
35、(2)在50ml烧瓶中加入500-600mg双-(2-甲氧基苯基)氧化膦,然后加入4-5ml的苯溶剂,接着加入0.3-0.5gkoh(50wt%)、70-80mg四丁基碘化铵和0.1-0.2ml1,3-二氯丙烷;将混合物搅拌加热到80-90℃下回流7-8h,然后冷却到室温;反应结束后,加入10 -20ml的蒸馏水淬灭,所得产物用20-30ml乙酸乙酯萃取3-5次;有机相加入无水硫酸钠干燥除水,过滤,旋蒸去除溶剂,所得到产物进一步过柱分离纯化,得到白色固体,即1,3-双(二(2-甲氧基苯基)氧化膦)丙烷。该步骤的反应式如式(2)。
36、
37、(3)首先在50ml的烧瓶中依次加入300-400mg1,3-双(二(2-甲氧基苯基)氧化膦)丙烷、0.5-1ml三氯硅烷(hsicl3)、1-2ml三丁胺(n-bu3n)和7-8ml的乙腈(ch3cn),混合物在70-80℃下搅拌5-6h,然后冷却至室温;反应结束后,加入2-3ml浓度为20%的naoh溶液淬灭,所得溶液用15-30ml乙酸乙酯提取3-5次;有机相加入无水硫酸钠干燥除水,过滤,旋蒸去除溶剂,所得到产物进一步过柱分离纯化,得到白色固体。该步骤的反应式如式(3)。
38、
39、(4)将带有搅拌子的100ml schlenk瓶连续抽真空并用红外灯烘烤1-2小时后冷却至室温,然后在氮气保护下,依次将0.2-0.5mmol制备好的二膦配体和0.2-0.5mmol pdcl2(cod)加入至20-40ml的ch2cl2溶剂中,在20-30℃常温下反应1-2h后,将反应混合物用冷肼抽滤到大约为5-10ml左右,随即添加20-40ml体积比为1-2:1-2的正戊烷与乙醚的混合物使得固体析出,将其滤出后,用正戊烷洗涤3-5次,然后常温真空下干燥,得到黄色固体,即所述二膦钯配合物。
40、本发明还提供一种上述二膦钯配合物在催化co和乙烯共聚生成可光降解聚酮的反应中的应用。
41、上述应用中,优选地,参与共聚反应的单体还包括丙烯。
42、上述应用中,优选地,所述可光降解聚酮包括二元极性聚酮和/或三元极性聚酮。
43、本发明还提供一种二元极性聚酮的制备方法,包括如下步骤:在上述上述二膦钯配合物和第一酸添加剂的作用下,使co和乙烯进行二元聚合反应,从而得到所述二元极性聚酮。
44、烯烃与极性单体(co)配位共聚反应主要受限于:(1)配位聚合催化剂的金属中心一般具有强lewis酸性,容易与极性单体发生σ-配位螯合作用,阻碍双键的π-配位,进而抑制单体的插入;(2)极性单体插入后,极性基团易于与金属中心配位形成稳定的螯合物;(3)极性单体插入后,形成的中间体易于发生链转移/链终止反应。因此在共聚反应中,co和钯金属中心很容易发生配位,得到高度稳定的co配合物,造成共聚的活性显著降低。对此,本发明采用合适的催化剂结构,得到对烯烃高活性的co配合物,提高共聚的活性。
45、上述二元极性聚酮的制备方法中,优选地,co和乙烯的投料压力比为1-5:1-5,更优选为2:3。
46、上述二元极性聚酮的制备方法中,优选地,二元聚合反应压力为20-80atm,更优选为20-60atm,反应温度为50-90℃,反应时间为1-5h。
47、上述二元极性聚酮的制备方法中,优选地,二膦钯配合物与co的摩尔比为0.5-1.5:1,更优选为0.93-1.03:1。
48、上述二元极性聚酮的制备方法中,优选地,所述第一酸添加剂与二膦钯配合物的摩尔比为50-100:1。
49、上述二元极性聚酮的制备方法中,优选地,所述第一酸添加剂选自自三氟乙酸、乙酸、高氯酸、磺酸、卤代羧酸、甲苯磺酸中的至少一种。
50、上述二元极性聚酮的制备方法中,优选地,在反应时,所述催化剂和第一酸添加剂溶解于溶剂中;所述溶剂选自甲醇、醋酸、硫酸、二甲基亚矾、n,n二甲基甲酰胺或水,更优选为甲醇。
51、上述二元极性聚酮的制备方法中,优选地,如图1所示,具体包括如下步骤:
52、高压反应釜真空下加热干燥,然后充入氮气保护,然后将计量好的二膦钯配合物以及第一酸添加剂都溶解在溶剂(如甲醇或者其混合溶剂)中,然后高压釜密封加热50-90℃搅拌均匀,先充入乙烯10-30atm,继续充入co至10-30atm,保持釜内压力20-60atm,聚合反应1-5h,反应完成后降温冷却,排气泄压,然后从高压釜内分离出聚合物,过滤,用丙酮洗涤至聚合物发白,然后真空干燥,得到共聚物,即所述二元极性聚酮。
53、本发明还提供一种上述二元极性聚酮的制备方法得到的二元极性聚酮(pok)。优选地,该二元极性聚酮的结构式为-(c2h4co)n-。
54、本发明还提供一种三元极性聚酮的制备方法,包括如下步骤:在上述二膦钯配合物和第二酸添加剂的作用下,使co、乙烯和丙烯进行三元聚合反应,从而得到所述三元极性聚酮。
55、co与乙烯在常规聚合中易形成完全交替共聚物,此类聚合物结构规整,结晶度高,在实际加工过程中加工难度大。本发明的应用通过加入第三单体丙烯,改善链段柔韧性,平衡聚酮综合性能,制备具有高催化剂活性、高分子质量、刚韧平衡的聚酮产品。
56、本发明的制备方法采用具有边臂效应的制备方法得到的二膦钯配合物或上述二膦钯配合物,在配体中引入磷原子和氧原子,通过边臂功能基团(磷原子和氧原子)与钯金属中心的协同作用,解决co与中心金属配位作用导致的聚合体系催化剂活性弱、产品分子量低等难题,增强乙烯与极性单体的共聚活性,提高催化剂活性,制备高分子量聚酮产品;此外,本发明还通过加入第三单体丙烯,改善链段柔韧性,平衡聚酮综合性能,制备具有高分子质量、刚韧平衡的三元极性聚酮产品。本发明合成三元极性聚酮的反应式如下:
57、
58、上述三元极性聚酮的制备方法中,优选地,co、乙烯和丙烯的投料压力比为1-20:1-20:1-10,更优选为1-10:1-10:1-5,进一步优选为2:2:1。
59、上述三元极性聚酮的制备方法中,优选地,三元聚合反应压力为30-80atm,反应温度为50-90℃,反应时间为1-5h。
60、上述三元极性聚酮的制备方法中,优选地,制备方法得到的二膦钯配合物或上述二膦钯配合物与co的摩尔比为0.5-1.5:1。更优选为0.93-1.03:1。
61、上述三元极性聚酮的制备方法中,优选地,所述第二酸添加剂与制备方法得到的二膦钯配合物或上述二膦钯配合物的摩尔比为50-100:1。
62、上述三元极性聚酮的制备方法中,优选地,所述第二酸添加剂选自三氟乙酸、乙酸、高氯酸、磺酸、卤代羧酸、甲苯磺酸中的至少一种。
63、上述三元极性聚酮的制备方法中,优选地,在反应时,所述二膦钯配合物和第二酸添加剂溶解于溶剂中;所述溶剂选自甲醇、醋酸、硫酸、二甲基亚矾、n,n二甲基甲酰胺或水,更优选为甲醇。
64、上述三元极性聚酮的制备方法中,优选地,如图1所示,具体包括如下步骤:
65、高压反应釜真空下加热干燥,然后充入氮气保护,然后将计量好的二膦钯配合物以及第二酸添加剂都溶解在溶剂(如甲醇或者其混合溶剂)中,然后高压釜密封加热50-90℃搅拌均匀,先充入乙烯10-30atm,继续充入co至10-30atm,再充如丙烯10-20atn并保持釜内压力30-80atm,聚合反应1-5h,反应完成后降温冷却,排气泄压,然后从高压釜内分离出聚合物,过滤,用丙酮洗涤至聚合物发白,然后真空干燥,得到共聚物,即所述三元极性聚酮。
66、本发明提供的技术方案,具有如下有益效果:
67、本发明的二膦钯配合物具有边臂效应,其通过边臂功能基团(磷原子和氧原子)与钯金属中心形成协同作用,克服因co与中心金属的配位作用导致的聚合体系催化剂活性弱、产品分子量低的问题,增强乙烯与极性单体(co)的共聚活性。本发明制备的具有边臂效应的二膦钯配合物的催化剂活性可达到10023.6kgpok/(gpd.h),共聚物重均分子量达到11.9×104,相对于普通配体1,3-二苯基磷基丙烷(dppp),其催化活性有了明显提升,所得共聚物的分子量也大大提高。
本文地址:https://www.jishuxx.com/zhuanli/20240619/1543.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。