一种丙环唑的合成方法与流程
- 国知局
- 2024-06-20 10:49:32
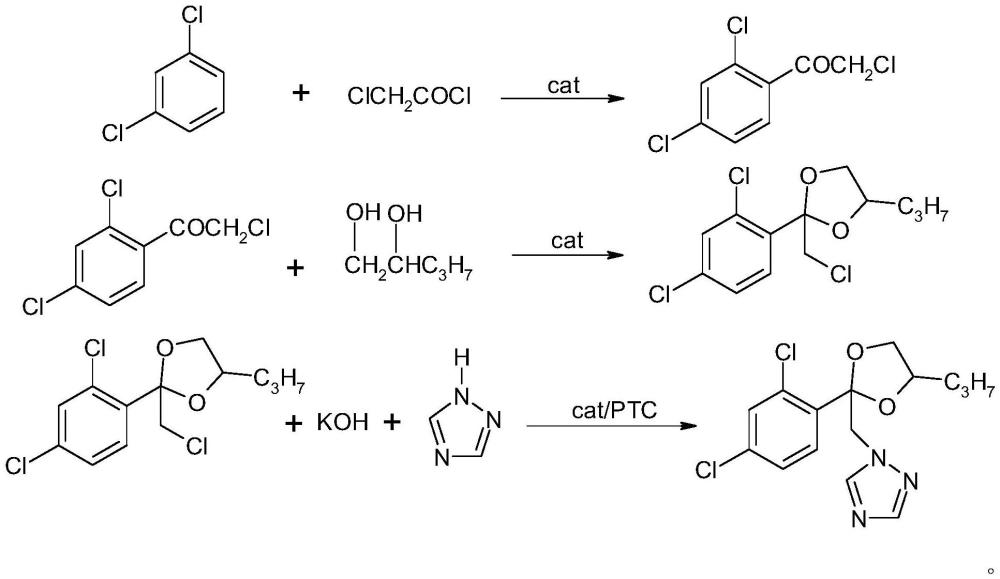
本发明涉及一种化合物的生产方法,具体涉及一种丙环唑的合成方法,属于化学合成。
背景技术:
1、丙环唑是一种具有保护和治疗作用的内吸性环菌唑类杀菌剂,其化学名称是1-[2-(2,4-二氯苯基)-4-丙基-1,3-二氧戊环-2-基甲基]-1h-1,2,4-三唑,是甾醇脱甲基化制剂,原药外观为淡黄色粘稠液体,沸点(13.3pa)180℃,比重(20℃)1.27g/cm3,在水中溶解度为110mg/l,易溶于有机溶剂。丙环唑对危害作物生长的多数真菌病害均有良好的防治效果,对子囊囊菌、担子菌和半知菌类等病原菌引起的病害具有良好的防治效果,其弥散剂用于大棚温室蔬菜、瓜、果病害的防治,持效期达3个月之久,丙环唑还具有一定的植物生长调节活性,具备增产、早熟、抗倒伏等多种功能。对人、畜低毒,无致畸、无突变作用。
2、目前所采用的工艺主要是以2,4-二氯苯乙酮、戊二醇、溴素、三氮唑等为原料,经环化、溴化、缩合、提纯得到丙环唑,其中环化生成的反应物在溴化过程易发生逆反应,导致转化率降低;溴化使用的液溴属于重大危险源,生产操作需要严格防护,且目前液溴价格持续增长,导致成本提高;缩合反应需先脱水合成三氮唑盐,反应时间长,物料粘稠,所得丙环唑需要经过高真空或硝酸成盐进行提纯。
技术实现思路
1、本发明所要解决的技术问题是针对现有技术中存在的不足,而提供一种丙环唑的合成方法,是一种以氯乙酰氯和间二氯苯为原料,经过傅克反应得到ω-氯-2,4-二氯苯乙酮,再与1,2-戊二醇进行环化反应得到2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷,通过在缩合反应中加入高效催化剂与相转移催化剂,提高2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与1,2,4-三氮唑的反应速率,一锅法合成丙环唑的生产工艺。通过选用氯乙酰氯做原料,避免了溴化反应步骤,避免了溴化反应所带来的成本高、危险性大等缺点,通过在缩合反应中加入高效催化剂与相转移催化剂,并使用固体碱进行合成,一锅法直接合成丙环唑,反应过程中无需脱水,操作简便,且有效避免丙环唑异构体的生成,反应收率高,副产物少,是一种清洁、高效的生产工艺。
2、为了实现上述目的,本发明采用如下技术方案:
3、一种丙环唑的合成方法,包括以下步骤:采用氯乙酰氯和间二氯苯在催化剂i的作用下反应得到ω-氯-2,4-二氯苯乙酮,ω-氯-2,4-二氯苯乙酮与1,2-戊二醇在催化剂ⅱ作用下进行环化反应得到2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷,然后再与1,2,4-三氮唑在溶剂中,与固体碱在催化剂ⅲ、相转移催化剂的作用下直接进行缩合反应得到丙环唑,化学反应式如下所示:
4、
5、上述技术方案中,所述的催化剂i为三氯化铝,间二氯苯与催化剂i的摩尔比为1:1-2。
6、上述技术方案中,所述的氯乙酰氯和间二氯苯反应中,间二氯苯与氯乙酰氯的摩尔比为1:1-1.5。
7、上述技术方案中,所述的氯乙酰氯和间二氯苯反应中,氯乙酰氯的滴加温度为0-20℃,滴加时间2-4hr,保温温度为20-40℃,保温时间为2hr。
8、上述技术方案中,所述的氯乙酰氯和间二氯苯反应中,反应在溶剂二氯乙烷中进行。
9、上述技术方案中,所述的催化剂ⅱ为对甲苯磺酸,ω-氯-2,4-二氯苯乙酮与催化剂ⅱ的质量比为1:0.01-0.05。
10、上述技术方案中,所述的ω-氯-2,4-二氯苯乙酮与1,2-戊二醇反应中,ω-氯-2,4-二氯苯乙酮与1,2-戊二醇的摩尔比为1:1.05-1.2。
11、上述技术方案中,所述的ω-氯-2,4-二氯苯乙酮与1,2-戊二醇反应中,环化反应温度为80-90℃,反应时间4-12hr。
12、上述技术方案中,所述的ω-氯-2,4-二氯苯乙酮与1,2-戊二醇反应中,反应在溶剂环己烷中进行。
13、上述技术方案中,所述的缩合反应中,2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与三氮唑的摩尔比为1:1-1.5。
14、上述技术方案中,所述的缩合反应中,溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮中的任意一种、两种及以上以任意比例混合而成的混合物。
15、上述技术方案中,所述的缩合反应中,固体碱为碳酸钠、碳酸钾、氢氧化钠、氢氧化钾中的任意一种、两种及以上以任意比例混合而成的混合物。
16、上述技术方案中,所述的缩合反应中,所述的固体碱为与三氮唑的摩尔比为0.9-1.1:1。
17、上述技术方案中,所述的缩合反应中,所述的催化剂ⅲ为碘化亚铜、氯化亚铜中的任意一种、两种以任意比例混合而成的混合物。
18、上述技术方案中,所述的缩合反应中,所述的2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与催化剂ⅲ的质量比1:0.001-0.05。
19、上述技术方案中,所述的缩合反应中,所述的相转移催化剂为四丁基溴化铵、四丁基氯化铵、十二烷基硫酸钠、冠醚的任意一种、两种及以上以任意比例混合而成的混合物。
20、上述技术方案中,所述的缩合反应中,所述的2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与相转移催化剂的质量比为1:0.005-0.05。
21、上述技术方案中,所述的缩合反应无需进行脱水,1,2,4-三氮唑与固体碱合成1,2,4-三氮唑盐后再与2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷反应合成丙环唑。
22、与现有技术相比,具有以下特点:
23、本发明采用氯乙酰氯与间二氯苯作原料,取消了正常工艺过程中的溴化工艺,避免了重大危险源溴的使用,降低了反应的危险性,在缩合反应过程中,在高效催化剂的作用下,2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与三氮唑在碱性条件下直接合成丙环唑,反应无需先脱水合成三氮唑盐后再进行反应。工艺条件温和,合成的丙环唑转化率高,有效避免丙环唑异构体的生成,副产物少,产品总收率、含量高,生产成本低,是一种清洁、高效的生产工艺。
技术特征:1.一种丙环唑的合成方法,其特征在于,包括以下步骤:采用氯乙酰氯和间二氯苯在催化剂i的作用下反应得到ω-氯-2,4-二氯苯乙酮,ω-氯-2,4-二氯苯乙酮与1,2-戊二醇在催化剂ⅱ作用下进行环化反应得到2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷,然后再与1,2,4-三氮唑在溶剂中,与固体碱在催化剂ⅲ、相转移催化剂的作用下直接进行缩合反应得到丙环唑,化学反应式如下所示:
2.根据权利要求1所述的合成方法,其特征在于,所述的催化剂i为三氯化铝,间二氯苯与催化剂i的摩尔比为1:1-2;所述的氯乙酰氯和间二氯苯反应中,间二氯苯与氯乙酰氯的摩尔比为1:1-1.5;所述的氯乙酰氯和间二氯苯反应中,氯乙酰氯的滴加温度为0-20℃,滴加时间2-4hr,保温温度为20-40℃,保温时间为2hr,反应在溶剂二氯乙烷中进行。
3.根据权利要求1所述的合成方法,其特征在于,所述的催化剂ⅱ为对甲苯磺酸,ω-氯-2,4-二氯苯乙酮与催化剂ⅱ的质量比为1:0.01-0.05;所述的ω-氯-2,4-二氯苯乙酮与1,2-戊二醇反应中,ω-氯-2,4-二氯苯乙酮与1,2-戊二醇的摩尔比为1:1.05-1.2;所述的ω-氯-2,4-二氯苯乙酮与1,2-戊二醇反应中,环化反应温度为80-90℃,反应时间4-12hr,反应在溶剂环己烷中进行。
4.根据权利要求1所述的合成方法,其特征在于,所述的缩合反应中,2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与三氮唑的摩尔比为1:1-1.5。
5.根据权利要求1所述的合成方法,其特征在于,所述的缩合反应中,溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮中的任意一种、两种及以上以任意比例混合而成的混合物。
6.根据权利要求1所述的合成方法,其特征在于,所述的缩合反应中,固体碱为碳酸钠、碳酸钾、氢氧化钠、氢氧化钾中的任意一种、两种及以上以任意比例混合而成的混合物;所述的缩合反应中,所述的固体碱为与三氮唑的摩尔比为0.9-1.1:1。
7.根据权利要求1所述的合成方法,其特征在于,所述的缩合反应中,所述的催化剂ⅲ为碘化亚铜、氯化亚铜中的任意一种、两种以任意比例混合而成的混合物;所述的2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与催化剂ⅲ的质量比1:0.001-0.05。
8.根据权利要求1所述的合成方法,其特征在于,所述的缩合反应中,所述的相转移催化剂为四丁基溴化铵、四丁基氯化铵、十二烷基硫酸钠、冠醚的任意一种、两种及以上以任意比例混合而成的混合物。
9.根据权利要求1所述的合成方法,其特征在于,所述的缩合反应中,所述的2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷与相转移催化剂的质量比为1:0.005-0.05。
10.根据权利要求1所述的合成方法,其特征在于,所述的缩合反应无需进行脱水,1,2,4-三氮唑与固体碱合成1,2,4-三氮唑盐后再与2-氯甲基-2-(2,4-二氯苯基)-4-丙基-1,3-二噁戊烷反应合成丙环唑。
技术总结本发明公开了一种丙环唑的合成方法,采用氯乙酰氯和间二氯苯在催化剂I的作用下反应得到ω‑氯‑2,4‑二氯苯乙酮,后再与1,2‑戊二醇在催化剂Ⅱ作用下进行环化反应得到2‑氯甲基‑2‑(2,4‑二氯苯基)‑4‑丙基‑1,3‑二噁戊烷,后再与1,2,4‑三氮唑在溶剂、固体碱、催化剂、相转移催化剂的作用下进行缩合反应得到丙环唑。本发明采用氯乙酰氯与间二氯苯为原料,节省了现有的溴化反应步骤,避免了溴化反应所带来的成本高、危险性大等缺点,在缩合反应时通过加入催化剂与相转移催化剂,提高了缩合反应速率,节省了三氮唑与碱脱水成盐的步骤,降低异构体及其它副产物的生成,提高了丙环唑的纯度,缩合反应收率>91%。技术研发人员:王明坤,孙淮成,谢邦伟,冯素流,唐伟受保护的技术使用者:江苏优嘉植物保护有限公司技术研发日:技术公布日:2024/6/18本文地址:https://www.jishuxx.com/zhuanli/20240619/404.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。