一种新型环孢素衍生物及其应用的制作方法
- 国知局
- 2024-09-19 14:28:36
本发明涉及一种新型环孢素衍生物,包含其的药物组合物,其制备方法,以及用于治疗由病毒感染、肿瘤、自身免疫、炎症类疾病治疗中的应用。
背景技术:
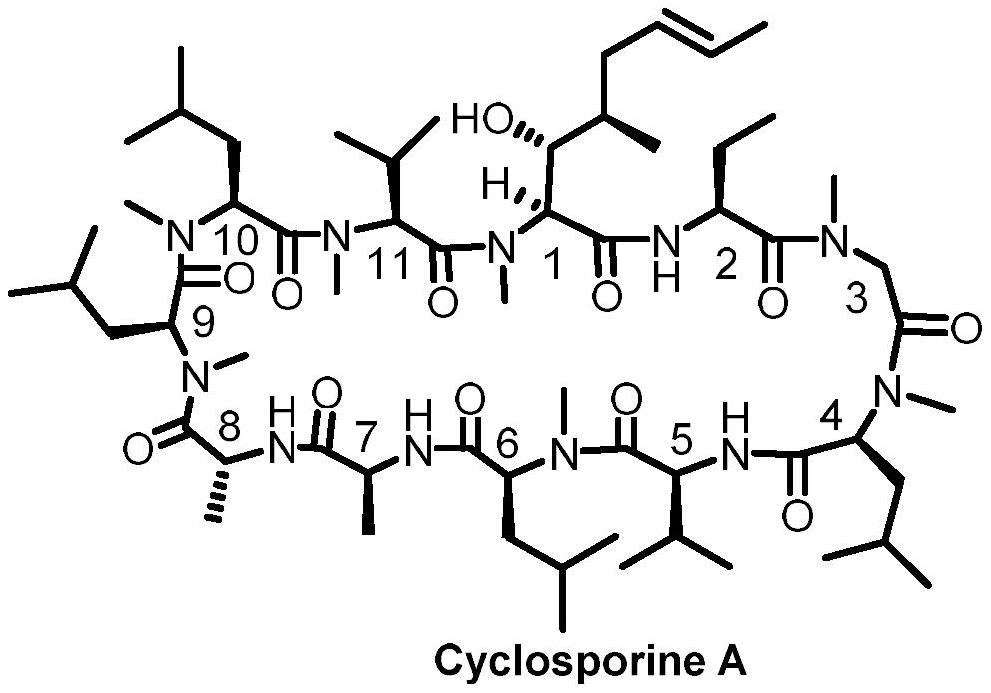
1、环孢素a(cyclosporine a,csa)是一种由土壤真菌中分离出的十一环肽,该环肽首位成环,该分子具有七个氮甲基取代及一个非天然氨基酸mebmt((4r)-4-((e)-2-butenyl)-4,n-dimethylthreonine)。环孢素a的结构如下所示:
2、
3、该分子的十一个氨基酸残基按顺序分别为:
4、一位氨基酸mebmt为:(4r)-4-[(e)-2-丁烯基]-4-甲基-l-苏氨酸((4r)-4-[(e)-2-butenyl]-4-methyl-l-threonine);
5、二位氨基酸α-abu为:l-α-氨基丁酸(l-α-aminobutyric acid);
6、三位氨基酸sar为肌氨酸(sarcosine);
7、四、六、九、十位氨基酸meleu为n-甲基-l-亮氨酸(n-methyl-l-leucine);
8、五位氨基酸val为l-缬氨酸(l-valine);
9、七位氨基酸ala为l-丙氨酸(l-alanine);
10、八位氨基酸d-ala为d-丙氨酸(d-alanine);
11、十一位氨基酸meval为n-甲基-l-缬氨酸(n-methyl-l-valine)。
12、环孢素a自1969年由瑞士的borel从土壤真菌中分离出以来,其在疾病治疗及药物研发中得到了广泛的应用。1983年环孢素a首先被fda批准用于治疗器官移植后的免疫排斥。随后机制研究证明环孢素的作用靶点为亲环素a(cyclophilin a,cypa),免疫抑制的作用机制为环孢素a与亲环素a及钙调蛋白形成三元复合物,该复合物的形成可以抑制钙调蛋白的磷酸化及t特别是th细胞的激活、增殖等。基于这一免疫抑制活性环孢素还被用于其他自身免疫类疾病,如牛皮癣、类风湿性关节炎、眼膜炎等免疫失调类疾病等,此外环孢素a的免疫调控作用还可以用于孕期妇女的保胎作用。
13、通过对csa进行结构改造,可以削弱其免疫抑制活性(钙调蛋白结合能力),保留或增强其cypa结合能力。基于这一原理,多个环胞霉素衍生物(alisporivir,scy-635,nim-811,stg-175)已经处于临床研究治疗丙肝及乙肝感染及癌症治疗中。环孢素类的亲环素抑制剂对多种人冠状病毒(hcov-229e,hcov-nl63,sars-cov,mers-cov)具有良好的抑制作用。其中alisporivir对于2019年开始出现的新冠病毒细胞抑制活性为ec50=0.46±0.04mm与对照药物chloroquine ec50=0.435±0.02mm的活性相当,但alisporivir表现出更低的细胞毒性(softic l,brillet r,berry f,ahnou n,nevers q,morin-dewaele m,hamadats,bruscella p,fourati s,pawlotsky j-m,ahmed-belkacem a.2020.inhibition ofsars-cov-2infection by the cyclophilin inhibitor alisporivir(debio 025).antimicrob agents chemother 64:e00876-20.)。目前,靶向病毒的药物治疗方案如,lopinavir联合ritonavir及chloroquine用于新冠病毒的临床方案未能改善病人症状,临床应用均以失败告终。而环孢素衍生物是通过靶向病毒复制所需的人体蛋白,不易产生抗药性。
技术实现思路
1、本发明通过设计、合成出一系列修饰后的环孢素衍生物,确定了如通式(i-0),特别是通式(i)及(ii)所示的化合物在细胞水平中对人冠状病毒hcov229e、sars-cov-2和hbv等病毒均呈现出较高的抑制活性。特别是,本研究中的示例化合物br-000514在细胞感染后表现出良好的抗sars-cov-2的活性,及很低的细胞毒性,可作为先导化合物为后续的研究奠定了基础。
2、本发明的目的在于提供通式(i-0)的化合物或其药学上可接受的盐,尤其是提供通式(i)和通式(ii)的化合物或其药学上可接受的盐。
3、根据本发明,如下通式(i-0)所示的化合物或其药学上可接受的盐:
4、
5、其中:
6、a选自:-ch2ch=chr1、-ch2ch2r1;
7、其中r1选自:
8、(a)-(ch2)n-ch3;
9、(b)-(ch2)p-cooh;
10、(c)-(ch2)q-nh(c=o)ch3;
11、(d)-苯基,所述苯基被以下基团一次或多次相同或不同地取代:-cooch3、-(ch2)nnh(c=o)ch3;优选地,所述苯基被以下基团一次或多次相同或不同地取代:-coome、-ch2nhac;
12、(e)-c3-c6环烷基;
13、b选自:-ch3、-ch2ch3、-ch(ch3)2;
14、c1和c2各自独立地选自:
15、(a)-h;
16、(b)5-7元杂环基、未取代或被三氟甲基取代的c6-c12芳基,优选地,所述芳基为苯基,所述杂环基为噻吩基,其中用于取代的取代基可以有一个或多个,例如1、2或3个;
17、(c)-chr21r22,r21和r22各自独立地选自:-h、-ch3、-ch2ch3、-ch2ch2ch3、噻吩基、未取代或被甲基、三氟甲基取代的苯基、ch3(ch2)n(c=o)o-;
18、(d)-c3-c6环烷基;
19、或者c1和c2一起形成=chr3,r3选自:
20、(a)-h;
21、(b)5-7元杂环基、未取代或被甲基、三氟甲基取代的c6-c12芳基,优选地,所述芳基为苯基,所述杂环基为噻吩基,其中用于取代的取代基可以有一个或多个,例如1、2或3个;
22、(c)-ch2r2,r2选自:-h、-ch3、-ch2ch3、-ch2ch2ch3;
23、(d)-c3-c6环烷基,例如所述环烷基为环丙基、环丁基;
24、优选地,r3选自:-h、-ch3、-ch2ch3、-ch2ch2ch3、噻吩基、未取代或被甲基、三氟甲基取代的苯基、-c3-c6环烷基;
25、c’选自:-h、-ch3、-ch2ch3、-ch2ch2ch3;
26、d1和d2各自独立地选自:-h、-ch2ch(ch3)2、-ch2c(oh)(ch3)2、-ch(ch3)(ch2ch3)、-ch(ch3)2,或者d1和d2与它们所连接的原子一起形成c3-c6饱和碳环;
27、或者a和d1或d2彼此连接形成长链桥环结构,所述长链桥环结构选自:-(ch2)n-ch=ch-(ch2)p-cr41r42-(c=o)nh-(ch2)q-、-(ch2)n-cr41r42-(c=o)nh-(ch2)n-,其中,r41和r42各自独立地选自:-h、-ch3;
28、n、p、q在每次出现时各自独立地选自:0、1、2、3、4、5。
29、根据本发明,通式(i-0)所示的化合物可由以下通式(i)所示:
30、
31、其中:
32、a选自:-ch2ch=chr1、-ch2ch2r1;
33、其中r1选自:
34、(a)-(ch2)n-ch3,其中n选自:0、1、2;
35、(b)-(ch2)p-cooh,其中p选自:1、2、3,优选地,p为3;
36、(c)-(ch2)q-nh(c=o)ch3,其中q选自:1、2、3,优选地,q为3;
37、(d)-苯基,所述苯基被以下基团一次或多次相同或不同地取代:-cooch3、-ch2nh(c=o)ch3;或-coome、-ch2nhac;
38、(e)-c3-c6环烷基,优选地,所述环烷基为环丙基;
39、b选自:-ch3、-ch2ch3、-ch(ch3)2;
40、c选自:
41、(a)-h;
42、(b)5-7元杂环基、未取代或被三氟甲基取代的c6-c12芳基,优选地,所述芳基为苯基,所述杂环基为噻吩基,其中用于取代的取代基可以有一个或多个,例如1、2或3个;
43、(c)-ch2r2,r2选自:-h、-ch3、-ch2ch3、-ch2ch2ch3;
44、(d)-c3-c6环烷基,例如所述环烷基为环丙基、环丁基;
45、c’选自:-h、-ch3、-ch2ch3、-ch2ch2ch3;
46、d选自:-ch2ch(ch3)2、-ch2c(oh)(ch3)2、-ch(ch3)(ch2ch3)、-ch(ch3)2。
47、优选地,在通式(i)的结构中,
48、a选自:-ch2ch=chr1、或-ch2ch2r1,
49、其中r1选自:
50、(a)-ch3;
51、(b)-(ch2)p-cooh;
52、(c)-(ch2)q-nh(c=o)ch3;
53、(d)-苯基,所述苯基被以下基团一次或多次相同或不同地取代:-cooch3、-ch2nh(c=o)ch3;或-coome、-ch2nhac;
54、(e)-c3-c6环烷基,所述环烷基为环丙基;
55、b选自:-ch3;
56、c选自:
57、(a)-h;
58、(b)5-7元杂环基、未取代或被三氟甲基取代的c6-c12芳基,优选地,所述芳基选自:苯基,所述杂环基选自:噻吩基,其中用于取代的取代基可以有一个或多个,例如1、2或3个;优选地,所述5-7元杂环基选自:噻吩基;所述未取代或被三氟甲基取代的c6-c12芳基选自:苯基、三氟甲基苯基;
59、(c)-ch2r2,r2选自:-h、-ch3、-ch2ch3、-ch2ch2ch3;
60、(d)-c3-c6环烷基,所述环烷基为环丙基、环丁基;
61、c’选自:-h、-ch3;
62、d选自:-ch2ch(ch3)2、-ch2c(oh)(ch3)2,
63、p、q分别如上所定义。
64、根据本发明,通式(i-0)所示的化合物可由以下通式(ii)所示:
65、
66、其中:
67、a选自:-ch2ch=chr1、-ch2ch2r1,
68、其中r1选自:
69、(a)-(ch2)n-ch3;
70、(b)-(ch2)p-cooh;
71、(c)-(ch2)q-nh(c=o)ch3;
72、(d)-苯基,所述苯基被以下基团一次或多次相同或不同地取代:-cooch3、-ch2nh(c=o)ch3;或-coome、-ch2nhac;
73、(e)-c3-c6环烷基,所述环烷基为环丙基;
74、b选自:-ch3、-ch2ch3、-ch(ch3)2;
75、c1选自:-h、-chr21r22,其中,r21和r22各自独立地选自:-h、-ch3、-ch2ch3、-ch2ch2ch3、噻吩基、未取代或被甲基、三氟甲基取代的苯基、ch3(ch2)n(c=o)o-,优选地,c1选自-h、-chr21r22,其中,r21和r22各自独立地选自:-h、-ch3、噻吩基、苯基、p-三氟甲基苯基、ch3(c=o)o-;
76、c’选自:-h、-ch3、-ch2ch3、-ch2ch2ch3;
77、d 1、d 2是相同或不同的且彼此独立地选自:-h、-ch2ch(ch3)2、-ch2c(oh)(ch3)2、-ch(ch3)(ch2ch3)、-ch(ch3)2;
78、或d1和d2与它们所连接的原子一起形成c3-c6饱和碳环,所述c3-c6饱和碳环任选地被取代基r4一次或多次相同或不同地取代,r4选自:-h、-ch3、-ch2ch3、-ch2ch2ch3;
79、或a和d1或d2彼此连接形成长链桥环结构,所述长链桥环结构选自:-(ch2)n-ch=ch-(ch2)p-cr41r42-(c=o)nh-(ch2)q-、-(ch2)n-cr41r42-(c=o)nh-(ch2)n-,其中,r41和r42各自独立地选自:-h、-ch3;
80、n、p、q如上通式(i-0)中所定义;
81、优选地,在通式(ii)的结构中,
82、a选自:-ch2ch=chr1、或-ch2ch2r1,
83、其中r1选自:
84、(a)-ch3;
85、(d)-苯基,所述苯基被以下基团一次或多次相同或不同地取代:-cooch3、-ch2nh(c=o)ch3;或-coome、-ch2nhac;
86、(e)-c3-c6环烷基,所述环烷基为环丙基;
87、b选自:ch3、-ch2ch3、-ch(ch3)2;
88、c1选自:-h、-chr21r22,其中,r21和r22各自独立地选自:-h、-ch3、噻吩基、苯基、p-三氟甲基苯基、ch3(c=o)o-;
89、c’选自:-h、-ch3;
90、d 1、d 2是相同或不同的且彼此独立地选自:-h、-ch2ch(ch3)2、-ch2c(oh)(ch3)2;
91、或d1和d2与它们所连接的原子一起形成c3-c6饱和碳环,所述c3-c6饱和碳环任选地被取代基r4一次或多次相同或不同地取代,r4选自:-h、-ch3、-ch2ch3、-ch2ch2ch3,优选地,所述c3-c6饱和碳环选自:环丙基;
92、或a和d1或d2彼此连接形成长链桥环结构,所述长链桥环结构选自:-(ch2)n-ch=ch-(ch2)p-cr41r42-(c=o)nh-(ch2)q-、-(ch2)n-cr41r42-(c=o)nh-(ch2)n-,其中,r41和r42各自独立地选自:-h、-ch3;
93、n、p、q如上所定义。
94、特别地,通式(i-0)所示的化合物如下所示:
95、
96、
97、
98、
99、
100、本发明的再一目的在于提供包含根据本发明的一种或多种上述通式(i-0)、(i)或(ii)的化合物或其药学上可接受的盐,和/或任选的药学上可接受的载体、赋形剂和/或稀释剂的药物组合物。
101、本发明的再一目的在于提供上述通式(i-0)、(i)或(ii)的化合物或其药学上可接受的盐,或者所述药物组合物用于制备亲环素抑制剂的用途。
102、本发明的再一目的在于提供上述通式(i-0)、(i)或(ii)的化合物或其药学上可接受的盐,或者所述药物组合物用于制备治疗或预防哺乳动物中亲环素介导的疾病或损伤的药物中的用途,尤其是可以用于制备治疗或预防病毒感染、癌症、自身免疫、炎症类疾病的药物中的用途。
103、本发明的再一目的在于提供一种用于抑制有需要的受试者中的亲环素的方法,所述方法包括对所述受试者施用有效抑制亲环素的量的一种或多种上述通式(i-0)、通式(i)或通式(ii)的化合物或其药学上可接受的盐,或所述药物组合物。
104、本发明的再一目的在于提供一种用于治疗或预防有需要的哺乳动物物种中的疾病的方法,所述方法包括对所述哺乳动物物种施用治疗有效量的一种或多种上述通式(i-0)、通式(i)或通式(ii)的化合物或其药学上可接受的盐,所述药物组合物,其中所述疾病选自病毒感染、癌症、自身免疫、炎症类疾病。
105、根据本发明,所述病毒包括冠状病毒、hbv,所述癌症包括鼻咽癌、宫颈癌、早幼粒白血病、肝癌、淋巴瘤;优选地,所述冠状病毒包括人冠状病毒、新型冠状病毒;所述鼻咽癌包括ebv阳性鼻咽癌、ebv阴性鼻咽癌。更优选地,所述人冠状病毒选自:hcov229e、所述新型冠状病毒选自:sars-cov-2。
106、本发明的有益效果:
107、在针对本发明的新型环孢素衍生物与cypa结合活性测定、人冠状病毒hcov229e抑制活性及细胞毒性测定、hbv抑制活性及细胞毒性测定、肿瘤细胞系的抗增殖活性实验(选取的细胞系分别为:人ebv阳性鼻咽癌细胞c666-1和hone1-ebv、人ebv阴性鼻咽癌细胞hk1和hone1-vc、人宫颈癌细胞hela、人早幼粒白血病细胞hl-60、人肝癌细胞huh7和人淋巴瘤细胞raji)、sars-cov-2抑制活性测定实验、以及br-000514在小鼠体内代谢研究等,充分证实本发明的化合物与csa相比,具有与cypa较高的结合活性,较强的人冠状病毒hcov229e抑制活性和/或较低的细胞毒性,hbv抑制活性测定和/或较低的细胞毒性;并且对于不同的肿瘤细胞系具有较为明显或突出的抑制活性。特别是br-000514在人冠状病毒sars-cov-2抑制活性ic50较磷酸氯喹(chloroquine phosphate)和瑞德西韦(remdesivir)还要更低,针对vero细胞毒性cc50>100μm,充分表明br-000514对人冠状病毒sars-cov-2具有优异的抑制活性,同时具有很低的细胞毒性。同时,br-000514在小鼠中代谢数据与环孢素csa在大鼠中代谢数据较为一致,充分表明该分子在体研究可以相对于环孢素更快的起效且一次服药可以具有更长的保护时间。故,本发明的新型环孢素衍生物具有优异的与cypa结合活性,并且对病毒感染、癌症有明显的抑制作用且细胞毒性较低。本发明的新型环孢素衍生物可用于制备治疗或预防哺乳动物中亲环素介导的疾病或损伤的药物中的用途。尤其可以用于制备治疗或预防病毒感染、癌症、自身免疫、炎症类疾病的药物中的用途。
本文地址:https://www.jishuxx.com/zhuanli/20240919/298290.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表