一种利用病毒宏基因组学检测犊牛肠道病毒的方法
- 国知局
- 2024-08-05 11:41:42
本发明涉及病毒的检测方法,具体涉及一种利用病毒宏基因组学检测犊牛肠道病毒的方法。
背景技术:
1、宏基因组学(metagenomics),也称为环境基因组学。通过针对土壤微生物群落组成及其功能进行分析,系统地将宏基因组学定义为:以环境样品中微生物群体的基因组为研究对象,以微生物多样性、种群结构、进化关系、功能活性及与环境之间的关系作为研究目的,通过功能基因筛选和测序分析进行的微生物群体研究。随着微生物学与分子生物学研究的不断深入,宏基因组学被确定为在微生物基因组学基础上发展起来的一种研究微生物遗传多样性和发现新类型微生物的新方法。目前,宏基因组学研究涵盖了海洋环境研究、植物和农业生物技术、人类遗传学、人与动物疾病诊断等多个研究领域。edwards与rohwer针对167个完整噬菌体基因组序列进行了系统发育与分类学统计归纳分析,提出噬菌体蛋白组树的分类方法,首次确定病毒宏基因组学概念为解析特定环境和生物样品中的病毒组成情况,进一步明确了未来主要研究方向包括建立dna病毒和rna病毒克隆与测序方法,深入研究基因水平转移与可移动遗传元件之间的关系,探索微生物基因组中未鉴定基因与病毒基因组之间的关系等。
2、病毒宏基因组学研究主要包括两种方法:泛病毒芯片(pan-viral microarrays)和高通量测序(high-throughput sequencing)。泛病毒芯片是将所有已知病毒代表性序列制成微阵列芯片,可以用来检测几乎所有已知病毒。vm研究在发展前期于环境微生物研究领域主要应用两种芯片,即功能基因芯片(geochip)和系统发育芯片(phylochip)。2003年,wang等根据genbank中各病毒属基因保守序列设计了含寡核苷酸探针的基因芯片鉴定了sars病毒。然而该技术仍然存在着无法鉴定未知新型病毒,且对于环境中丰度低、变异大的病毒进行检测时效果不理想的技术缺陷。随着基因测序技术不断发展,高通量测序技术极大程度上拓宽了对新病毒和已知病毒变异情况的检测范围,逐渐成为当前vm研究的主流技术。基于高通量测序技术建立的vm不依赖于传统的分子生物学方法,以特定环境中所有病毒群落的基因组为研究对象,通过对特定环境中的所有病毒进行富集,随后对病毒核酸进行高通量测序,并通过生物信息学方法与已知病毒基因序列进行相似性比对,从而了解环境样品中病毒组构成,研究过程主要包括四个部分:样品处理、文库构建、高通量测序、生物信息学分析。
3、目前病毒宏基因组检测还存在一些缺陷,例如1)病毒在样本中的含量较低往往难以支持高通量测序导致检测结果不够全面,需要进行样本和核酸富集;2)常规方法使用0.22μm滤膜过滤细菌,然而目前存在部分病毒直径大于0.22μm,利用0.22μm滤膜过滤样本液可能导致病毒漏检;3)样本液游离宿主核酸干扰检测结果;4)illumina原始测序数据存在低质量数据和与病毒无关核酸数据影响后续组装准确性,需要剔除无关核酸数据后进行分析等问题,因此本发明欲开发一种基于宏基因组学实现对犊牛肠道病毒检测的方法。
技术实现思路
1、本发明的目的是提供一种利用病毒宏基因组学检测犊牛肠道病毒的方法,明确样本中犊牛肠道病毒群落组成,为分析我国东北地区犊牛病毒性腹泻病原及其遗传变异的趋势提供了有效的数据支持。
2、为了达到上述目的,本发明提供了一种利用病毒宏基因组学检测犊牛肠道病毒的方法,其特征在于,包含以下步骤:
3、(1)样本中全基因组的提取;
4、(2)对提取的全基因组进行文库构建;
5、(3)对构建的文库进行高通量测序;
6、(4)对测序结果进行生物信息学分析。
7、进一步地,上述步骤(1)中全基因组的提取为dna和rna全基因组的提取。
8、进一步地,上述步骤(2)中的文库构建包含:将rna样品片段化,反转录制备成cdna并连接接头,同时纯化接头连接产物;对纯化的产物进一步扩增并纯化,用于提高测序文库浓度。
9、进一步地,上述的连接接头为对cdna片段进行末端补平修复,在其3’端添加碱基“a”转换为黏性末端。
10、进一步地,上述的扩增为桥式pcr扩增,具体包含如下:
11、其中,pcr反应体系为50μl,包含纯化后的接头连接产物20.0μl,2×pcr mix 25.0μl,5primer 2.5μl,i7 primer 2.5μl;
12、pcr反应程序为98℃预变性45s;98℃10s,60℃15s,72℃30s,共15次循环;72℃终延伸1min,4℃保存。
13、进一步地,上述步骤(3)中的高通量测序为:通过illumina测序平台进行高通量测序,每个测序样品产生6g数据量。
14、进一步地,上述步骤(4)中的生物信息学分析包含:
15、(1)将测序结果通过bbmap v38.51软件与ncbi数据库进行比对,筛选过滤掉相应的rrna、宿主和细菌相关reads,从而获得有效读长序列(clean reads);
16、(2)使用kraken 2软件与ncbi nt库、viral refseq库进行blast比对以完成cleanreads的分类注释,确定进化关系最接近的候选参考序列,并确定比对序列所属的种属;
17、(3)使用拼接软件spades v3.13.0和soapdenovo对clean reads进行从头组装成contig,组装根据de brujin算法设置k-mer长度为21、33、55等多个参数进行,综合评定组装结果,选取最佳k-mer组装结果,从而获得基因组序列;
18、(4)将拼接组装得到的所有contig与ncbi数据库进行blast比对,并基于比对结果进行物种鉴定,提取注释为病毒的基因序列进行遗传进化分析,通过iq-tree确定最适核苷酸替换模型并基于贝叶斯法构建系统发育树。
19、本发明的检测方法具有以下优点:
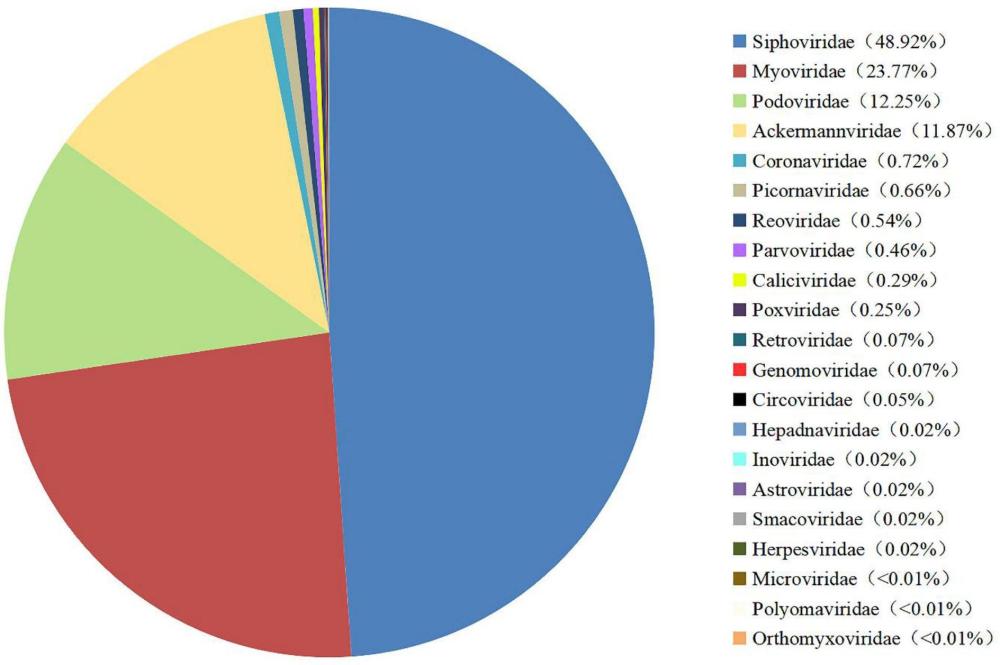
20、本实验利用病毒宏基因组学方法结合vm技术,对大庆周边牛场的粪便样品中病毒性病原进行了测序,明确了大庆周边犊牛肠道病毒群落组成。检测到btov、bkv、bnov以及bnev等新发犊牛腹泻病毒。组装出brv、bcov、boastv相关contig序列,并针对此三种病毒与我国其他流行株进行了遗传进化分析。为分析我国东北地区犊牛病毒性腹泻病原及其遗传变异的趋势提供了有效的数据支持。
技术特征:1.一种利用病毒宏基因组学检测犊牛肠道病毒的方法,其特征在于,包含以下步骤:
2.根据权利要求1所述的方法,其特征在于,所述步骤(1)中全基因组的提取为dna和rna全基因组的提取。
3.根据权利要求1所述的方法,其特征在于,所述步骤(2)中的文库构建包含:将rna样品片段化,反转录制备成cdna并连接接头,同时纯化接头连接产物;对纯化的产物进一步扩增并纯化,用于提高测序文库浓度。
4.根据权利要求3所述的方法,其特征在于,所述的连接接头为对cdna片段进行末端补平修复,在其3’端添加碱基“a”转换为黏性末端。
5.根据权利要求3所述的方法,其特征在于,所述的扩增为桥式pcr扩增,具体包含如下:
6.根据权利要求1所述的方法,其特征在于,所述步骤(3)中的高通量测序为:通过illumina测序平台进行高通量测序,每个测序样品产生6g数据量。
7.根据权利要求1所述的方法,其特征在于,所述步骤(4)中的生物信息学分析包含:
技术总结本发明公开了一种利用病毒宏基因组学检测犊牛肠道病毒的方法,包含以下步骤:1)样本中全基因组的提取;2)对提取的全基因组进行文库构建;3)对构建的文库进行高通量测序;4)对测序结果进行生物信息学分析。通过本发明提供的检测方法可以对样本的病毒进行遗传进化分析,包括已知的病毒和新发现的病毒株,可以明确样本中犊牛肠道病毒群落组成,为分析我国东北地区犊牛病毒性腹泻病原及其遗传变异的趋势提供了有效的数据支持。技术研发人员:赵建军,李玉铎,陈天杰受保护的技术使用者:黑龙江八一农垦大学技术研发日:技术公布日:2024/8/1本文地址:https://www.jishuxx.com/zhuanli/20240802/258856.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。