一种从纳米孔测序数据定量检测DNA修饰的方法
- 国知局
- 2024-10-21 14:57:17
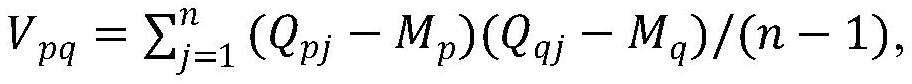
本发明涉及生物医学,具体涉及一种检测dna修饰的方法。
背景技术:
1、纳米孔分析技术起源于coulter计数器的发明以及单通道电流的记录技术。生理与医学诺贝尔奖获得者neher和sakamann在1976年利用膜片技术测量膜电势,研究膜蛋白及离子通道,推动了纳米孔测序技术的实际应用进程。1996年,kasianowicz等提出了利用a-溶血素对dna测序的新设想,是生物纳米孔单分子测序的里程碑标志。随后,mspa孔蛋白、噬菌体phi29连接器等生物纳米孔的研究报道,丰富了纳米孔分析技术的研究。li等在2001年开启了固态纳米孔研究的新时代。
2、利用纳米孔研究dna等生物分子是一种崭新的生物技术,与以往的dna测序技术相比纳米孔测序技术无需各种酶的参与、也无需对dna进行化学修饰、标定物插入等生物或化学处理,直接采用物理方法读出dna序列,检测成本较低。由于组成dna的四种碱基腺漂吟(a)、鸟漂吟(g)、胞(c)和胸腺喀呢(t)的分子结构及体积大小均不同,单链dna(ssdna)在核酸外切酶的作用下被迅速逐一切割成脱氧核糖核昔酸分子,当单个碱基在电场驱使下通过纳米级的小孔时,不同碱基的化学性质差异导致穿越纳米孔时引起的电流的变化幅度不同,从而得到所测dna的序列信息。
3、目前用于测序的纳米孔可以分为三类:生物纳米孔、固态纳米孔和生物-固态杂化纳米孔。第一,生物纳米孔是天然的生物纳米器件,具有特定的孔径结构、生物活性及能够插入脂双分子层膜的能力,如a-溶血素(ahl)纳米孔。ahl是目前最广泛使用的生物纳米孔的分析物质,由293个氨基酸多肽构成,可插入到纯净的双分子层脂膜中形成蘑菇状七聚体,组装成跨膜通道;ahl七聚体纳米孔主要由帽型区(cap,入口cis端直径为2.6nm)、边缘区(rim,直径为1.4nm)和主干区(stem,入口trans端直径为2.2nm)三部分构成。ahi纳米孔永久开通不关闭,耐强酸和强碱,高温、高电压下较稳定。第二,纳米孔固态纳米孔主要是在氮化硅、二氧化硅和石墨烯等绝缘材料上用离子刻蚀技术、电子刻蚀技术、聚焦电子束(feb)或离子束(fib)等制作出的微小孔洞。目前固态纳米孔的制备,首先用常规微加工技术制作30~500nm厚的悬空薄膜,再用离子束或电子束等在硅或其他材料薄膜表面钻出2~100nm的孔洞。dna检测中所需的纳米孔直径都是1~2nm,可在前述研究的基础上,进一步采用沉淀物质收缩、离子束辐射、电子束辐射等收缩技术减小纳米孔的尺寸,从而达到更小目标尺寸的纳米孔。第三,生物固态杂化纳米孔即将生物纳米孔固定在固态的膜上,例如通过化学方法将一个长链dsdna的末端与α-溶血素结合在一起,可以通过电泳使其进入到氮化硅纳米孔中形成同轴,呈一条线的结构,实现dna的测序。
4、测序前,需要制备样本,即对dna链进行接头处理。dna测序可分为1dread和2dread两种。1dread即只测序模板链,而2dread模板链和互补链均进行测量。接头序列分为三种,分别为蓝色的头部接头序列(lead adaptor)、红色的发夹接头序列(hairpinadaptor)以及棕色的尾部接头序列(trailing adaptor)。在1dread测序中,leadadaptor和trailingadaptor分别连接到dna片段的两端。接头序列有助于纳米孔上的蛋白质分子补获该测序链,并确保测序序列沿着单向在毫秒尺度沿链位移在2dread测序中,发夹接头序列通过共价连接将一个链连接到另一个链上,将双链分子的两个链进行连续排序。开始测序时,头部接头序列带领测序分子进入纳米孔,先对模板链进行测序,然后发夹接头序列进入纳米孔,之后对互补链进行测序,最后尾部接头序列通过。利用pairwisealignment,将结果组合成2dread。
5、其中2dread测序具有八个不同阶段,第一阶段,蛋白质分子在薄膜上开口,形成纳米孔;第二阶段,纳米孔捕获dna链的头部接头序列;第三阶段,头部接头序列通过纳米孔;第四阶段,模板链通过纳米孔;第五阶段,发夹接头序列通过纳米孔;第六阶段,互补链通过纳米孔;第七阶段,尾部接头序列通过纳米孔;第八阶段,纳米孔恢复状态;之后依次通过头部接头序列、模板链、发夹接头序列、互补链、尾部接头序列;每一个接头序列都有其独特的电流信号模式,从而辅助将模板链和互补链区分开。
6、每个纳米孔连接到四个wells,每次从其中的一个well产生数据,不同纳米孔产生reads的能力是不同的。纳米孔中产生的电流由传感器每秒测量数千次,之后被传送到设备的专用集成电路上进行处理。在minion的碱基识别流程中,原始电流被处理并分割成一系列“events”,每个事件具有此段电流信号的平均值、方差以及持续时间等。处理后的电流数据再传送到主机中名为minknow的软件上,miknow软件利用基于维特比算法的隐马尔可夫模型(hmm)搜索最优路径产生测序序列,并将该序列和对应的events等相关诠释信息生成原始fast5文件。该原始fast5文件还会被上传到amazon云中,基于云的metrichor会根据电流值进行进一步碱基识别,最后原始fast5的数据以及metrichor返回的碱基再次识别数据组合成最终fast5文件,该文件可被用户下载到指定目录中。每个纳米孔测序的单链数据对应一个fast5文件,即一个fast5文件对应一组读段(read)。由于包含测序原始电信号,因此fast5文件非常庞大,难以存储及传输,不利于公共数据存储库中共享。
7、dna修饰在调控基因表达、维持基因组稳定性、产生与维持基因印记、x染色体沉默等生物过程中扮演着重要角色,同时也是癌症等疾病的重要标志物。dna修饰包括5-甲基胞嘧啶(5mc)、5-羟甲基胞嘧啶(5hmc)、5-甲酰基胞嘧啶(5fc)、5-羧基胞嘧啶(5cac)、n6-甲基腺嘌呤以及n4-甲基胞嘧啶(4mc)等许多种类型。纳米孔测序是检测dna修饰的革命性平台,因为其无需亚硫酸氢盐处理或免疫沉淀等样品预处理即可直接检测dna修饰。纳米孔测序之所以可以直接检测dna修饰,是因为dna修饰在测序过程中得以保留,并对测序仪的原始电压信号产生影响。因此在理论上即使纳米孔测序数据是为了研究基因组结构变异等其他目的而产生的,也有可能被重新利用来研究dna修饰。然而在实践中现有的方法很难实现重利用大多数纳米孔测序数据,由于数据量过大等原因,大多数公开数据或大规模纳米孔测序数据不保留原始信号。目前已有的从纳米孔测序数据中检测dna修饰的方法都需要利用包含原始测序信号的fast5文件或由fast5文件导出的包含dna修饰信息的文件。这些方法从原始信号当中提取特征,比较修饰的碱基与未修饰的碱基之间差别,利用机器学习方法区分两者实现自动化的dna修饰检测。因此,现有技术很难利用公开的纳米孔测序数据或者大规模纳米孔测序数据检测dna修饰。
技术实现思路
1、本发明针对现有技术的不足,提出了一种新的从纳米测序数据中定量检测dna修饰的方法,这种新方法命名为nanofreelunch,该方法能够无需原始信号fast5文件,仅利用碱基识别(basecalling)之后包含测序dna序列以及碱基测序质量的fastq文件中即可实现dna修饰的定量检测,即检测每个基因组位点有多少被测序的碱基包含修饰。
2、第一方面,本发明提供一种定量检测dna修饰中提取数据特征的方法,进一步的,所述特征提取是从测序错误率以及测序质量分数这两大类特征中提取每个潜在包含dna修饰的位点周边测序质量分数的一到四阶矩、两两位点的联合错误率。
3、所述方法包括如下步骤:
4、s1在n个读段完全覆盖某cpg位点周边区域中,将基因组第i个位置的来自第j个读段的碱基记作rij,测序质量分数记为qij,参考基因组第i个位置的碱基记为ti。其中i=-m,...,m,j=1,...,n。测序质量分数的一阶矩(均值向量)为(1)所示
5、mean(q)=[mi]i∈[-m,m] (1)
6、其中是一个维数1×(2m+1)的行向量。
7、s2测序质量分数的二阶矩(协方差矩阵)为(2)所示
8、cov(q)=[vpq]p,q∈[-m,m] (2)
9、其中cov(q)是一个维数(2m+1)×(2m+1)的矩阵。
10、s3测序质量分数的三阶矩(联合偏度)为(3)所示
11、coskewness(q)=[spq]p,q∈[-m,m] (3)
12、其中σp与σq是标准差。coskewness(q)是一个维数为(2m+1)×(2m+1)的矩阵。
13、s4测序质量分数的四阶矩(联合峰度)为(4)所示
14、coskurtosis(q)=[kpq]p,q∈[-m,m] (4)
15、其中σp与σq为标准差。coskurtosis(q)是一个维数为(2m+1)×(2m+1)的矩阵。
16、s5两两位点的联合测序错误率为
17、error(r)=[estpq]s,t∈[c,c,g,t,,d],p,q∈[-m,m] (5)
18、其中i(·)为指示函数,d为删除,error(r)是一个维数为5×5×(2m+1)×(2m+1)的数组。
19、s6上述得到的一到四阶矩和两两位点的联合错误率为定量检测dna修饰中提取的特征数据。
20、第二方面,本发明提供一种从纳米孔测序数据定量检测dna修饰的方法,所述方法包括如下步骤:
21、s01序列比对:使用三代测序序列比对软件如minimap2将fastq文件包含的测序读段向参考基因组做序列比对;
22、s02特征提取:以第一方面所述特征提取方法得到测序质量分数的一到四阶矩、两两位点的联合错误率;
23、s03机器学习模型的构建:以上述提取的特征为预变量,输入机器学习算法,以guppy预测的dna修饰水平为响应,通过相关软件建立gradient boosting(gb)模型;
24、s04模型效能评估:利用human pangenome的hg01109纳米孔测序数据作为训练数据,ont open data中的gm24385数据和human pangenome的其他数据作为测试数据评估nanofreelunch的性能。
25、进一步的,所述序列比对是获得覆盖该区域从上游到下游碱基经过序列比对的读段。
26、进一步的,所述模型构建,优选xgboost软件的juliaapi(版本1.5.2)训练建立gradient boosting(gb)模型,其中学习步长“eta”设为0.1,回归树数量“num_round”设为200,回归树深度“max_depth”为6,其余参数为默认。
27、第三方面,本发明提供一种定量检测dna修饰的模型,所述模型包括数据处理、模型训练以及dna修饰水平预测三大模块,所述数据处理包括序列比对和特征提取;所述模型训练模块以“已知”dna修饰水平为响应以及特征提取模块提取的特征作为输入训练gb模型;所述dna修饰水平预测模块是利用训练好的gb模型对基因组位点上的dna修饰水平进行定量预测。
28、进一步的,所述序列比对是获得覆盖该区域从上游到下游碱基经过序列比对的读段。
29、进一步的,所述特征提取是基于第一方面所述的特征提取方法获得测序质量分数的一到四阶矩、两两位点的联合错误率。
30、进一步的,所述模型构建,优选xgboost软件的juliaapi(版本1.5.2)训练建立gradient boosting(gb)模型,其中学习步长“eta”设为0.1,回归树数量“num_round”设为200,回归树深度“max_depth”为6,其余参数为默认。
31、第四方面,本发明上述1-3方面在检测dna修饰中的应用。
32、进一步的,所述dna修饰包括5-甲基胞嘧啶(5mc)、5-羟甲基胞嘧啶(5hmc)、5-甲酰基胞嘧啶(5fc)、5-羧基胞嘧啶(5cac)、n6-甲基腺嘌呤以及n4-甲基胞嘧啶(4mc)等。
33、进一步的,所述应用包括基因印记和/或dna甲基化与组蛋白修饰的关联分析中的应用。
34、第五方面,一种测序错误率和/或测序质量分数在定量检测dna修饰中提取数据特征方法中的应用。
本文地址:https://www.jishuxx.com/zhuanli/20241021/319905.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表