一种基于雌激素受体-表观遗传因子互作网络的乳腺癌蛋白标志物筛选方法
- 国知局
- 2024-11-06 15:09:34
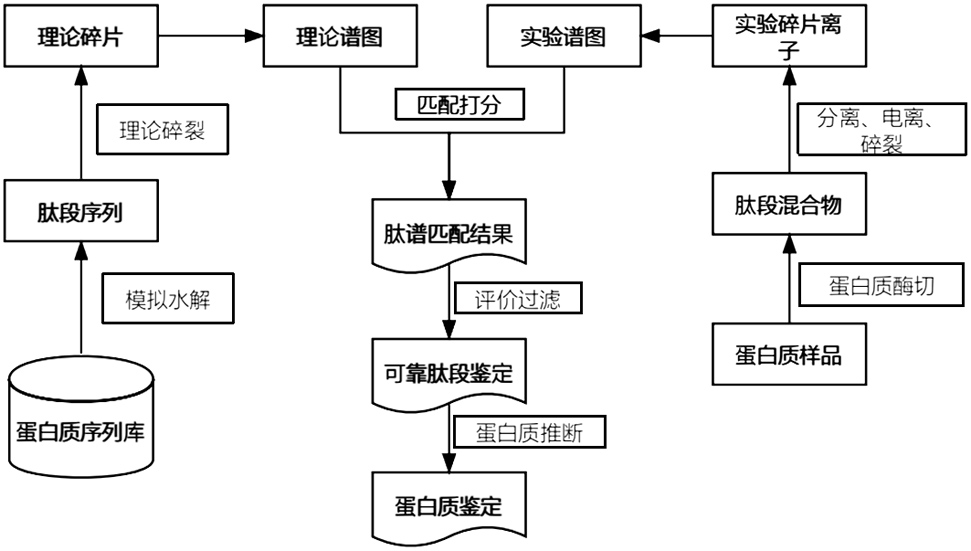
本发明提供一种基于雌激素受体-表观遗传因子互作网络的乳腺癌蛋白标志物筛选方法,属于乳腺癌蛋白标志物筛选。
背景技术:
1、乳腺癌作为一种常见的女性恶性肿瘤,表现出高度的异质性,包括分子亚型、临床病理特征和治疗反应等方面的差异;在临床实践中,进行准确分型与预后判断对于个体化治疗策略的选择至关重要,目前的乳腺癌分型方法往往依赖于基因组学和转录组学等技术,然而这些方法忽略了蛋白质层面的变化,尤其是雌激素受体及其互作蛋白在乳腺癌发生发展中的作用,导致分类的准确性不高;此外,传统的乳腺癌标志物检测技术大多局限于单一分子的研究,存在标志物的特异性不足、筛选流程繁琐导致检测效率低等问题,缺乏对乳腺癌细胞内部蛋白质相互作用的综合考虑。
技术实现思路
1、本发明为了克服现有技术中存在的不足,所要解决的技术问题为:提供一种基于雌激素受体-表观遗传因子互作网络的乳腺癌蛋白标志物筛选方法的改进。
2、为了解决上述技术问题,本发明采用的技术方案为:一种基于雌激素受体-表观遗传因子互作网络的乳腺癌蛋白标志物筛选方法,包括如下的筛选步骤:
3、步骤一:通过挖掘和分析蛋白质组学数据,构建一个全面的雌激素受体与表观遗传因子相互作用网络:
4、步骤1.1:从公共数据库proteomexchange、uniprot、ncbi中收集多组雌激素受体erα/β和表观遗传因子mettl3/14、tet2的蛋白互作组质谱数据,应用maxquant软件对蛋白质组进行定性和定量分析,进行肽段搜库匹配;
5、步骤1.2:使用基于二项式分布概率的算法对肽段-谱图进行匹配评分,进一步通过评分对肽段进行排序,确定肽段修饰:
6、首先计算理论碎片质量与质谱中峰的匹配数量 k,然后通过计算至少有 k个匹配的概率,并取其负对数乘以10来得到对应谱图的分数s( q, loss),该分数反映理论碎片质量列表与质谱质量列表之间的相似性,计算公式为:
7、;
8、式中: n表示理论离子的总数, k表示理论碎片质量与质谱中峰的匹配数量, q表示质量区间内允许出现的峰的个数,用于计算单个随机匹配的概率;
9、在质谱分析中,化合物分子经历碎裂过程,生成不同的碎片离子,产生中性损失,每次都会计算包含中性损失和排除中性损失的分数,选择两个分数中的最大值,并对于来自反向数据库的序列采用相同的评分操作,计算公式为:
10、;
11、式中: s( q)表示包含中性损失和排除中性损失的分数中的最大值,通过计算不同q值下的分数,选择最佳分数;
12、基于上述评分操作,从蛋白质列表中筛选出核受体与表观遗传因子;
13、步骤1.3:搜集蛋白互作数据库string和biogrid中已发表的雌激素受体与表观遗传因子互作证据,将结果与步骤1.2中筛选得到的核受体与表观遗传因子进行整合,使用cytoscape软件绘制蛋白质互作网络图,绘制的蛋白质互作网络图包括雌激素受体与核受体以及各种表观遗传修饰因子之间的相互作用;
14、步骤1.4:针对乳腺癌蛋白质组数据,使用limma包软件将乳腺癌样本与癌旁正常组织样本进行差异对比分析,筛选出在乳腺癌组织中差异表达的蛋白,这些差异蛋白与乳腺癌的发生和发展有关;
15、步骤二:运用机器学习方法确定乳腺癌新分型,然后筛选出蛋白标志物:
16、步骤2.1:基于一致性聚类算法确定乳腺癌新分型:
17、在差异蛋白中筛选出与雌激素受体互作的核受体和表观遗传因子,作为分类和标志物的候选者,利用这些差异蛋白在乳腺癌样本中的表达量数据,采用一致性聚类算法对乳腺癌进行无监督聚类;
18、采用中心点划分的pam算法进行无监督聚类,通过选择代表性的样本作为中心点,将数据集中的样本划分到与其最近的中心点中,找到最优的中心点配置,使得所有样本与其对应的中心点之间的距离总和最小;
19、步骤2.2:基于蛋白质组数据设计集成分类器模型pec,将乳腺癌样本中蛋白表达量作为特征,将带有分类标签的乳腺癌样本作为目标变量,融合多个单独的分类器预测并筛选蛋白质标志物,集成分类器模型pec具体的设计方法为:
20、采用网格搜索法,通过穷举参数空间中的所有可能组合,确定每个分类器的最佳参数,进而优化单独分类器的性能,在此过程中,采用软投票策略,将每个分类器的预测结果进行加权平均,其中权重取决于每个分类器的置信度;
21、将各个单独分类器组合成一个投票分类器,将多个分类器的预测结果结合起来,确定最终的分类决策。
22、所述步骤1.1中进行肽段搜库匹配的具体方法为:
23、步骤1.1.1:样本制备与蛋白质提取;
24、步骤1.1.2:利用聚丙烯酰胺凝胶电泳分离蛋白质,根据等电点将蛋白质分离,再根据其分子量进一步分离,得到蛋白质图谱;
25、步骤1.1.3:对蛋白质进行消化,使用胰蛋白酶将蛋白质水解成肽段,再使用质谱仪对消化后的蛋白质进行分析,采集肽段质谱数据;
26、步骤1.1.4:将上述实验测得的肽段质谱与数据库中的理论肽段质谱进行匹配,肽段质谱的匹配策略使用肽段匹配指标,通过比对肽段质谱中的质谱峰与数据库中的理论质谱峰,来确定蛋白质的鉴定结果;
27、步骤1.1.5:通过蛋白质免疫印迹实验检测蛋白表达的水平以及蛋白相互作用,验证质谱分析鉴定到的蛋白质,利用生物学实验验证鉴定到的蛋白质在生物学过程中的功能和作用。
28、所述步骤2.1中采用的pam算法的具体计算步骤为:
29、步骤2.1.1:初始化:共有n个样本,随机选择k个样本作为中心点;
30、步骤2.1.2:计算距离和分配样本:计算每个样本与中心点之间的距离,将每个样本分配到与其最近的中心点的类别中;
31、步骤2.1.3:更新中心点:对于第i个类中除了中心点以外的所有其他点,按顺序计算每个样本与其他样本的距离之和,即准则函数的值,计算公式为:
32、;
33、式中:e表示误差的平方和,即所有点到它们所属聚类中心距离的平方和,表示聚类中心数量,即聚类的类别数,p表示数据集中的一个点,即一个数据样本,oi表示第i个分类 c i的中心点,dist(p, oi)表示数据点p到聚类中心oi的距离;
34、步骤2.1.4:迭代更新:重复进行分配样本和更新中心点的过程,直到收敛为止,即各个中心点不再改变或已经达到设定的最大迭代次数;
35、步骤2.1.5:输出结果:得到最终的聚类中心数量。
36、所述步骤2.2中设计集成分类器模型pec对数据处理的具体方法为:
37、步骤2.2.1:输入蛋白质在样本中的表达矩阵,其中每行代表一个样本,每列代表一个蛋白质的表达量,分类信息列代表每个样本所属的类别;
38、步骤2.2.2:从输入的数据中分离出特征矩阵x和标签向量y,划分训练集和测试集,将70%的数据作为训练集,30%的数据作为测试集;
39、步骤2.2.3:初始化模型:使用网格搜索法,对于每组参数组合,在训练集上使用10折交叉验证,选择预设的参数组合作为最终模型的参数;
40、步骤2.2.4:将初始化的各个模型组合成一个投票分类器,采用软投票策略;
41、步骤2.2.5:使用训练集数据对投票分类器进行拟合;
42、步骤2.2.6:输出蛋白标志物贡献度值:
43、投票分类器对各个初始化模型的预测结果进行加权投票,得出最终的预测结果;
44、遍历投票分类器中的每个模型,提取其特征重要性,计算所有模型特征重要性的平均值,得到每个特征的平均贡献度;
45、对于计算蛋白在不同分组中的贡献度,使用投票分类器对训练集进行预测,得到每个样本属于各个类别的概率,将这些概率作为每个特征在不同分组中的贡献度。
46、本发明相对于现有技术具备的有益效果为:本发明为解决目前乳腺癌分型与标志物筛选方法中存在的缺陷,通过构建雌激素受体-表观遗传因子互作网络,综合考虑乳腺癌细胞内部蛋白质相互作用,提高分型的准确性,优化标志物的筛选过程,本发明能够更精确地识别乳腺癌中的关键蛋白质标志物,有效提高筛选效率和准确性,为乳腺癌后续的精准医疗提供有力支持。
本文地址:https://www.jishuxx.com/zhuanli/20241106/325589.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。