DHCR24抑制化合物的制作方法
- 国知局
- 2024-11-21 11:54:14
本发明涉及适用于δ24-脱氢胆固醇还原酶(δ24-dehydrocholesterolreductase,dhcr24)的抑制,特别是dhcr24的选择性抑制的化合物。这些化合物用作药物,特别是用作用于治疗和/或预防dhcr24介导的病症(例如非酒精性脂肪性肝炎(non-alcoholic steatohepatitis,nash)、动脉粥样硬化性心血管疾病(atheroscleroticcardiovascular disease,ascvd)或多发性硬化(multiple sclerosis,ms))的药剂。
背景技术:
1、已知胆固醇是重要的储存脂质和细胞构建物质。在过去的十年中,对胆固醇生物合成及其生物合成前体的生物学作用的理解发生了显著的变化。胆固醇生物合成与多种不同的疾病有关,并且研究集中在参与胆固醇生物合成的关键中间体和酶的生物学功能。
2、胆固醇生物合成分为前甲羟戊酸途径和后角鲨烯途径,后者也称为远端胆固醇生物合成。胆固醇从头生物合成是由甲羟戊酸途径(从乙酰辅酶a开始)中的十一种酶和参与远端胆固醇生物合成的九种酶完成的。后者进一步分为bloch途径和kandutsch-russell途径(参见图1)。
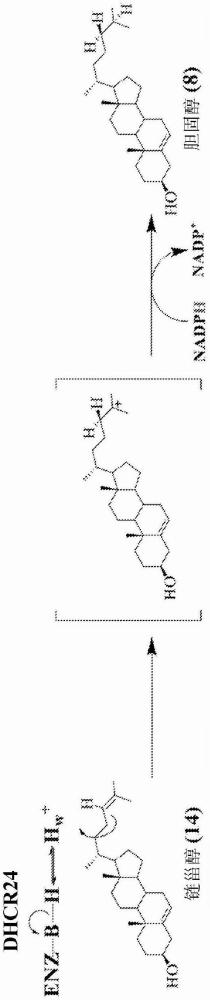
3、bloch途径包含δ24-不饱和中间体,并通过酶dhcr24的作用与kandutsch-russel分支相互连接。dhcr24是膜结合酶,其催化前体甾醇侧链中δ24-双键的厌氧还原(参见图1和图2)。已经表明,两种途径并未被严格分开,而是观察到两种途径的组织和细胞类型特异性相互作用,其中偏爱kandutsch-russel途径。dhcr24的主要底物是羊毛甾醇(4,4,14-三甲基胆甾-8,24-二烯-3β-醇,参见图1中的(1))和酵母甾醇(胆甾-8,24-二烯-3β-醇,参见图1中的(11)),以及胆甾-7,24-二烯-3β-醇(参见图1中的(12))。
4、δ24双键的还原可在bloch途径的最后步骤发生,其将链甾醇(胆甾-5,24-二烯-3β-醇,参见图1和2中的(14))转化为胆固醇(参见图1和2中的(8))。除nadph以外,dhcr24不需要辅因子。δ24双键的还原分两步进行,通过在c24处最初引入质子,在c25处产生阳离子高能中间体(high energy intermediate,hei),随后由nadph亲核加成氢化物(参见图2)。dhcr24的功能障碍或抑制导致哺乳动物胆固醇的生物合成通过bloch途径进行,最终导致链甾醇的积累。
5、链甾醇病(desmosterolosis)(mim 602398)是影响dhcr24基因的罕见遗传性病症。链甾醇病是非常罕见的疾病,仅有少数临床上描述的病例。链甾醇病伴有严重异常,例如小头畸形伴胼胝体发育不全、抽搐、眼球震颤、斜视和小颌畸形。已经发现,链甾醇的轻度积累对生活力没有影响,尤其是与富含胆固醇的饮食结合时,如dhcr24突变的杂合携带者所例示的。因此,通过抑制dhcr24在体内适度积累链甾醇被证明是无毒的。单个等位基因上dhcr24突变的携带者已被证明具有正常的胆固醇水平,其中链甾醇的血浆浓度仅提高了1.5倍。
6、在生物学上,dhcr24的作用是多种多样的,并且dhcr24的抑制是用于治疗多种疾病的有前景的药物靶点。因此,需要具有选择性、强效和无毒的dhcr24抑制剂,其在许多治疗性领域可以是有用的。
技术实现思路
1、在第一方面中,本发明提供了式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药。式(10)化合物为:
2、
3、其中:
4、g是选自以下的稠环体系:
5、
6、b选自:
7、
8、r1选自氢、-c(=o)r6和c1-6烷基;
9、r2选自氢和c1-6烷基;
10、r3选自由式(2)表示的基团、-[c(r7)2]n-x、-ch=cr6r7、-cr7=n-n(r6)2、5元至10元杂芳基和5元至10元杂环烷基,其中5元至10元杂芳基和5元至10元杂环烷基任选地被选自-oh、卤素、-cn、-nh2、-no2、c1-6烷基和c1-6烷氧基的一个或更多个基团取代;
11、
12、n是1至6的整数;
13、w选自o和nr6;
14、y选自氢和c2烯基,其中c2烯基任选地被一个或更多个卤素取代;
15、x选自卤素、-oh、-sh、-o-z、-s-z、-s-s-z、5元至10元杂芳基和5元至10元杂环烷基,其中5元至10元杂芳基和5元至10元杂环烷基任选地被选自-oh、卤素、-cn、-nh2、-no2、c1-6烷基和c1-6烷氧基的一个或更多个基团取代;
16、每个r4独立地选自氢和c1-6烷基;
17、r5选自氢和c1-6烷基;
18、每个r6独立地选自氢和c1-6烷基;
19、每个r7独立地选自h、c1-6烷基和c2-6烯基,其中c1-6烷基和c2-6烯基任选地被一个或更多个卤素取代;
20、z选自c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基,其中c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基任选地被选自-oh、卤素、-cn、-nh2、-no2、c1-6烷基和c1-6烷氧基的一个或更多个基团取代。
21、式(10)化合物不是以下之一:
22、
23、
24、式(10)中的虚线表示由g表示的稠环体系的位置。类似地,g的一个结构中的虚线代表由b表示的基团的位置。
25、发明人出乎意料地鉴定了一类抑制dhcr24的化合物。这些化合物是dhcr24的选择性、强效和无毒抑制剂。鉴于dhcr24的多种生物学作用,化合物对抑制dhcr24的选择性对于其在治疗或预防dhcr24介导的病症中的药物或临床用途是重要的。本发明化合物对dhcr24的抑制提高了内源性链甾醇水平。
26、与dhcr24的其他抑制剂相比,通过本发明鉴定的化合物在配制用于临床用途的药物产品时具有有利的特性(例如,可应用lipinski规则)。因此,化合物具有提高的水溶性和更好的稳定性。预期这些化合物也适用于直接经口施用。
27、本发明的另一个方面提供了式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药,其用于治疗或预防dhcr24介导的病症。本发明的这一方面还提供了治疗或预防dhcr24介导的病症的方法。该方法包括向对象施用式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药。
28、在第二方面中,本发明提供了式(1)化合物,或者其可药用盐、溶剂合物、水合物或前药,其用于治疗或预防dhcr24介导的病症。
29、本发明的第二方面还提供了治疗或预防dhcr24介导的病症的方法。该方法包括向对象施用式(1)化合物,或者其可药用盐、溶剂合物、水合物或前药。
30、在本发明的第二方面中,式(1)化合物为:
31、
32、其中:
33、g是选自以下的稠环体系:
34、
35、b选自:
36、
37、r1选自氢、-c(=o)r6和c1-6烷基;
38、r2选自氢和c1-6烷基;
39、r3选自由式(2)表示的基团、-[c(r7)2]n-x、-c(=o)nhor8、-c(=o)or8、-cr7=n-n(r6)2、-ch=cr6r7、c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基,其中c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基任选地被选自-oh、卤素、-cn、-nh2、-no2、c1-6烷基和c1-6烷氧基的一个或更多个基团取代;
40、
41、n是1至6的整数;
42、w选自o、s和nr6;
43、y选自氢、c1-6烷基、c2-6烯基、氨基-c1-6烷基、(单-c1-6烷基氨基)c1-6烷基和(二-c1-6烷基氨基)c1-6烷基,其中c1-6烷基和c2-6烯基任选地被一个或更多个卤素取代;
44、x选自卤素、-oh、-sh、-n(r6)2、-o-z、-s-z、-s-s-z、-c(=o)r7、c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基,其中c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基任选地被选自-oh、卤素、-cn、-nh2、-no2、c1-6烷基和c1-6烷氧基的一个或更多个基团取代;
45、每个r4独立地选自氢和c1-6烷基;
46、r5选自氢和c1-6烷基;
47、每个r6独立地选自氢和c1-6烷基;
48、每个r7独立地选自h、c1-6烷基和c2-6烯基,其中c1-6烷基和c2-6烯基任选地被一个或更多个卤素取代;
49、r8选自c1-6烷基和c2-6烯基,并且
50、z选自c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基,其中c6-10芳基、5元至10元杂芳基和5元至10元杂环烷基任选地被选自-oh、卤素、-cn、-nh2、-no2、c1-6烷基和c1-6烷氧基的一个或更多个基团取代。
51、式(1)中的虚线表示由g表示的稠环体系的位置。类似地,g的一个结构中的虚线代表由b表示的基团的位置。
52、本文中呈现的实验数据,特别是体内数据,提供了令人信服的证据:该化合物可在临床上用于治疗或预防dhcr24介导的病症。这些化合物可在脑内产生作用(例如,其不被血脑屏障阻止)。
53、本发明的另一个方面是提供药物组合物。该药物组合物包含如本文中所限定的式(1)或(10)化合物,或者其可药用盐、溶剂合物、水合物或前药,以及可药用赋形剂。
54、在另一个方面中,本发明提供了如本文中所限定的式(1)或式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药,或者如本文中所限定的药物组合物,其用于治疗和/或用作药物。
55、在另一个方面中,本发明提供了式(1)或式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药在制备用于治疗或预防dhcr24介导的病症的药物中的用途。
56、本发明的另一个方面提供了体内或体外抑制dhcr24活性的方法。该体内或体外方法包括使细胞与式(1)或式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药,或者如本文中所限定的药物组合物接触。或者,该体内方法可包括向对象施用式(1)或式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药,或者如本文中所限定的药物组合物。
57、在另一个方面中,本发明提供了组合,其包含式(1)或式(10)化合物,或者其可药用盐、溶剂合物、水合物或前药,以及一种或更多种另外的治疗剂。
58、本发明的任一特定方面的优选的、适当的和任选的特征也是任何其他方面的优选的、适当的和任选的特征。
本文地址:https://www.jishuxx.com/zhuanli/20241120/333305.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表