一种光介导无催化构建C(sp3)–C(sp3)键化合物的方法
- 国知局
- 2025-01-10 13:17:25
本发明属于有机光合成,具体涉及一种光介导无催化构建c(sp3)–c(sp3)键化合物的方法。
背景技术:
1、烷基取代基可以增加药物分子的亲脂性,增加药物分子的立体性并提高与蛋白的相互作用,在药物发现中至关重要。因此开发简单且稳定的c(sp3)–c(sp3)键构建策略在有机合成领域一直是一项紧迫的任务。传统的金属催c(sp3)–c(sp3)偶联(例如ni和pd)通常需要具有定位基团的烷烃、化学计量的氧化剂或其他添加剂。此外,还需要对亲电试剂(例如烷基卤化物)进行预官能团化,操作步骤繁琐。因此需要开发高效简洁的烷烃直接c(sp3)-h键官能化方法具有重要意义。
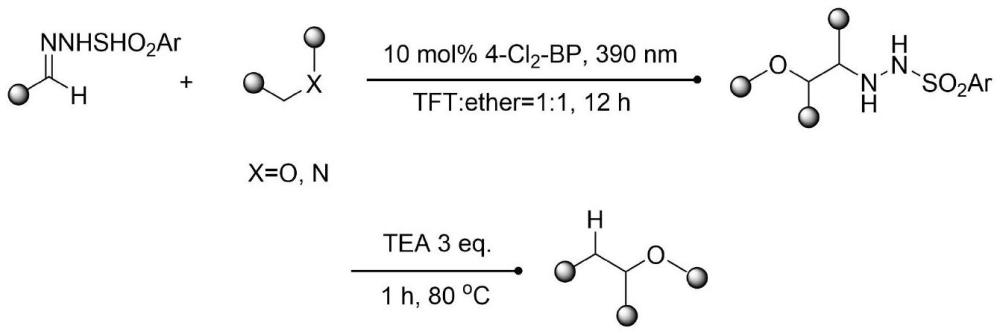
2、近年来,随着光学化学合成的快速发展,由macmillan、hashmi和martin团队报道的可见光介导的光催化氢原子转移(hat)策略与镍催化相结合,成功实现c(sp3)-h键直接烷基化。为了提高c(sp3)–c(sp3)键构建的可持续性,等人报道了一种两步合成策略,首先通过hat转移过程,随后在80℃下与三乙胺(tea)作用将n-磺酰腙与醚或胺结合,反应方程式表示如下:
3、
4、为了扩大n-磺酰腙在构建c(sp3)–c(sp3)键中的应用,课题组在光氧化还原条件下引入了以羧酸衍生酯为自由基前体的脱氧交叉亲电偶联,通过两步反应,成功实现c(sp3)–c(sp3)键构建;反应方程式表示如下:
5、
6、除上述方法外,n-甲苯磺酰腙可以与金属催化剂形成稳定且安全的金属卡宾中间体,随后进行c-h插入,这一反应机制在c(sp3)–c(sp3)偶联中得到应用。scott和decicco、noels、davies和dias报道了受体和受体/供体卡宾形成随后进行c-h插入这一重大进展。bi等在该领域中取得了重要进展,该团队在贵金属银催化和加热条件下分别实现n-三氟醚基腙与烷烃或醚偶联,反应方程式表示如下:
7、
8、尽管可见光催化技术在有机合成中得到了广泛应用,但目前极少有研究关注于无催化剂的条件下,利用可见光直接将烷烃/醚与烷基化亲电试剂相结合的策略。这种策略的原子经济性更高,且成本较低,操作简单,因此开发无催化剂且无过渡金属参与的c(sp3)–c(sp3)偶联方法具有显著的经济和应用价值。
技术实现思路
1、本发明基于上述研究,提供了一种安全、绿色的光介导无催化策略实现c(sp3)–c(sp3)化合物的高效构建。在惰性气体保护下,以对甲苯磺酰腙衍生物和非活化的烷烃/醚为原料,碱和有机溶剂存在下,在光源下照射反应,分离纯化后得到c(sp3)–c(sp3)偶联化合物;本发明能够缓慢释放供体/供体重氮化合物,光解生成单线态卡宾中间体,随后通过三元环过渡态选择性地插入到烷烃或醚的c-h键中;原料廉价易得,操作简单,绿色环保,为构建c(sp3)–c(sp3)提供了一条新途径,同时该方法还可以应用于克级反应及药物分子的后期修饰。
2、为了实现上述目的,本发明所采用的技术方案如下:
3、本发明以酮、对甲基苯磺酰肼、非活化的烷烃或醚和碱为原料,或直接以对甲苯磺酰腙衍生物、非活化的烷烃或醚、碱为原料通过无催化光介导策略得到产物,实现c(sp3)–c(sp3)的构建。两种原料合成c-h插入产物的方式基本相同,仅采用第一种原料时,先进行酮、对甲基苯磺酰肼间的密封搅拌,然后与非活化的烷烃或醚进行c(sp3)–c(sp3)的偶联过程。
4、本发明所述一种光介导无催化构建c(sp3)–c(sp3)的方法,包括如下步骤:碱存在时以对甲苯磺酰腙衍生物1和烷烃或醚2为原料,在光照下反应得到c(sp3)–c(sp3)偶联化合物3;反应方程式如下:
5、
6、其中:r1和r2各自独立选自h、c1-4烷基、苄基及其取代物、苯并呋喃及其取代物、苯并噻吩及其取代物、吲哚及其取代物、吡啶及其取代物、苯基及其取代物、萘基及其取代物;取代物为c1-4烷基、c1-4烷氧基、c1-4烷氧羰基烷氧基、亚甲基二氧基、boc、卤素、苯氧基;r3、r4、r5、r6和r7各自独立选自h、烷基;或r3、r4、r5一起通过碳原子组成环状结构;r6和r7通过碳原子组成环状结构。
7、进一步地,在上述技术方案中,所述对甲苯磺酰腙衍生物1典型结构为:
8、
9、进一步地,在上述技术方案中,所述非活化烷烃/醚2的典型结构为:
10、
11、进一步地,在上述技术方案中,所述碱选自dbu、dbn、dabco、cs2co3、k2co3、k3po4、koh、naoh、nah或t-buok中的一种或多种。优选为dbn、k2co3或cs2co3。
12、进一步地,在上述技术方案中,所述有机溶剂选自二氯甲烷、乙腈、氯仿、氟苯、二氯乙烷、烷烃或醚溶剂中的任意一种或多种组合。优选为烷烃或醚溶剂。
13、进一步地,在上述技术方案中,对甲苯磺酰腙衍生物1、非活化烷烃/醚2与碱摩尔比为1:1-500:1-6。
14、进一步地,在上述技术方案中,反应温度为20-35℃;反应时间为4-20小时。
15、进一步地,在上述技术方案中,光源波长为400-460nm;光源距离反应管为2-6厘米。
16、进一步地,在上述技术方案中,反应在惰性氛围下进行。
17、进一步地,在上述技术方案中,所述对甲苯磺酰腙衍生物1采用羰基化合物(醛或酮)与对甲苯璜酰肼在醇类溶剂中反应得到。其中,典型的醛或酮结构如下:
18、
19、进一步地,在上述技术方案中,后处理操作为:反应结束,采用乙酸乙酯和水萃取反应产物,无水硫酸钠干燥后旋除溶剂,通过柱层析分离纯化,得到c(sp3)–c(sp3)构建化合物。
20、本发明的有益效果
21、1、本发明以酮、对甲基苯磺酰肼和非活化的烷烃或醚或者对甲苯磺酰腙衍生物与非活化的烷烃或醚为主要原料,具有原料刺激性小、易于获得的特点。
22、2、本发明反应关键为通过缓慢释放供体/供体重氮物质,光解生成单线态卡宾中间体,随后通过三元环过渡态选择性地插入到烷烃或醚的c-h键中。
23、3、反应过程在常温常压下,利用光介导无催化反应构建c(sp3)–c(sp3)化合物,不仅合成工艺简单,显著提高合成产率,而且反应后对环境污染小,绿色环保。
24、4、原料廉价易得,操作简单,绿色环保,为实现c(sp3)–c(sp3)键的构建提供了一种可持续且易于获取的合成方法,该方法还可应用于克级合成和药物分子的后期修饰。
技术特征:1.一种光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于,包括如下步骤:碱存在时以对甲苯磺酰腙衍生物1和烷烃或醚2为原料,在光照下反应得到c(sp3)–c(sp3)偶联化合物3;反应方程式如下:
2.根据权利要求1所述光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于:所述碱选自dbu、dbn、dabco、cs2co3、k2co3、k2po4、koh、naoh、nah或tbuok中的一种或多种。
3.根据权利要求1所述光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于:所述有机溶剂选自二氯甲烷、乙腈、氯仿、氟苯、二氯乙烷、烷烃或醚溶剂中的任意一种或多种组合。
4.根据权利要求1所述光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于:对甲苯磺酰腙衍生物1、烷烃或醚2与碱摩尔比为1:1-500:1-6。
5.根据权利要求1所述光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于:反应温度为20-35℃;反应时间为4-20小时。
6.根据权利要求1所述光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于:光源波长为400-460nm;光源距离反应管为2-6厘米。
7.根据权利要求1-6任意一项所述光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于:反应在惰性氛围下进行。
8.根据权利要求1所述光介导无催化构建c(sp3)–c(sp3)键化合物的方法,其特征在于:对甲苯磺酰腙衍生物1采用羰基化合物(醛或酮)与对甲苯磺酰肼在非活化烷烃或醚类溶剂中缩合反应得到。
9.根据权利要求1所述光介导无催化构建c(sp3)–c(sp3)键的方法,其特征在于:
技术总结本发明公开了一种光介导无催化构建C(sp3)‑C(sp3)键的方法,属于有机光合成技术领域。在惰性气体保护下,以对甲苯磺酰腙衍生物1和非活化烷烃或醚2为原料,在碱和有机溶剂中存在下,将反应管在427nm光源下照射4‑20小时,分离纯化后得到C(sp3)‑C(sp3)键偶联化合物3;本发明反应关键为通过缓慢释放供体/供体重氮化合物,光解生成单线态卡宾中间体,随后通过三元环过渡态选择性地插入到烷烃或醚的C‑H键中;该反应体系原料廉价易得,操作简单,绿色环保,为实现C(sp3)‑C(sp3)键的构建提供了一种绿色高效且易于操作的合成策略。同时该方法还可以应用于药物分子的克级合成和后期修饰,展示了其良好的实用性。技术研发人员:张卫东,张宇,王金鑫,李倩楠,张娜,王丁刚,卓苗苗受保护的技术使用者:上海中医药大学技术研发日:技术公布日:2025/1/6本文地址:https://www.jishuxx.com/zhuanli/20250110/352189.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表