一种α-取代手性膦酸酯类化合物的合成方法
- 国知局
- 2024-06-20 11:32:28
本发明属于化学合成,具体涉及一种α-取代手性膦酸酯类化合物的合成方法。
背景技术:
1、作为α-芳基丙酸的类似物,对映纯的α-取代膦酸如萘普生、布洛芬和膦胺霉素等,具有十分广泛的生物活性,在生物学、药学和化学领域具有重要研究价值。过去几十年里,研究人员在其不对称合成方法开发中付出了巨大努力。其中,最为广泛报道的合成方法是ru,rh,ir,ni等过渡金属催化(tmc)的烯基膦酸酯不对称氢化反应(图1,a)。然而,tmc方法通常需要在高压氢气气氛下使用昂贵且有害的金属催化剂,而且每种底物一般都需要用特定配体以获得最佳选择性,并一般只适用于乙烯基磷酸酯底物。另一种较为有效的方法,是通过手性h-亚磷(膦)酸酯与α,β-不饱和醛、酮、酯以及硝基烷烃的不对称pudovik反应(图1,b)。但是,具有磷手性的h-亚磷(膦)酸酯原料实际上需要提前制备。2019年,fu课题组报道了一种镍/双噁唑啉催化,烯烃与α-溴代膦酸酯亲电体的还原加氢烷基化反应,但需使用2.5当量的(meo)3sih和cs2co3(图1,c)。此外,亲核性碳负离子与烯基膦酸酯的不对称加成反应也有相关报道(图1,d),但由于可得含磷底物相对较少,因此鲜有广泛应用。
2、通过可以回收利用的手性辅基诱导作用,是一种较为传统但却非常可靠的磷原子α-碳不对称官能化的策略方法,具有底物易得、操作简单、立体选择性高和原子经济性等优点。如光活性的1,2-氨基乙醇、1,2-二胺、薄荷醇以及binol等,已被成功用于膦酸酯毗邻碳手性中心的构建中(图1,e)。但是遗憾的是,上述已知手性诱导反应仍然存在立体选择性低和应用范围窄等问题。总之,如何在磷原子邻近碳原子上,有效构建具有不同烷基或芳基官能团的碳手性中心,仍然具有较大的技术挑战性。
技术实现思路
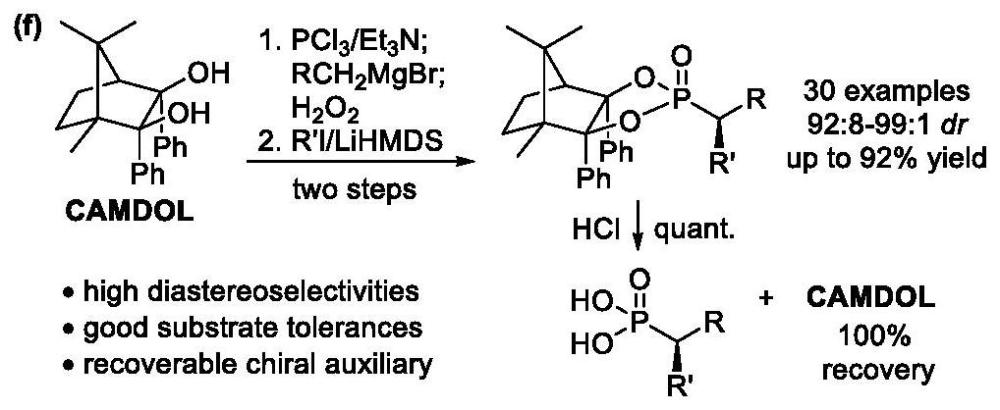
1、本方法提供了一种通过两步反应,快速高效合成α-取代手性膦酸酯类化合物的方法。
2、第一方面,本发明提供一种α-取代手性膦酸酯类化合物的合成方法,反应式如下式i:
3、
4、反应式中,r为甲基、异丙基、环己基、苯基、2-甲氧基苯基、3-甲氧基苯基、4-甲氧基苯基、4-氟苯基、4-氯苯基、4-异丁基苯基、6-甲氧基-2-萘基;r'为甲基、乙基、庚基、异丙基、异丁基、2-氰基乙基、2-氟乙基、2-二氟乙基、2-三氟乙基、烯丙基、环丙基甲基、环己烯基、苄基、2-甲基苄基、3-甲基苄基、4-甲基苄基、4-氯苄基、4-碘苄基、4-甲氧基苄基、2-喹啉基;
5、所述合成方法反应步骤分为两步,具体如下:
6、步骤一:将樟脑二醇加入反应瓶中,用惰性气体置换后,用四氢呋喃溶解,在-5~5℃环境下加入三乙胺和三氯化磷,搅拌并充分反应;然后,加入格氏试剂至原料反应完毕;再加入过氧化氢氧化,继续搅拌;反应完全后,加入饱和氯化铵淬灭,萃取后柱层析纯化,得到中间产物1;
7、步骤二:将中间产物1加入反应瓶中,用惰性气体置换后,用甲苯溶解,在-85~-70℃加入lihmds,搅拌并充分反应,然后加入碘代烷继续反应;反应完全后加入饱和氯化铵淬灭,萃取后柱层析纯化,得到α-取代手性膦酸酯。
8、优选地,所述樟脑二醇、三氯化磷、三乙胺、格氏试剂、过氧化氢、lihmds、碘代烷的摩尔比为1:1.1:2.5:1.1:1.1:1.5:1.5。
9、优选地,所述步骤一、二中,用惰性气体置换的具体操作为用氮气置换三次。
10、优选地,所述步骤一中,在0℃加入三乙胺和三氯化磷,搅拌10-20min;加入格氏试剂至原料反应完毕;加入过氧化氢氧化,继续搅拌30min。
11、优选地,所述步骤二中,在-78℃加入lihmds,搅拌30min,然后加入碘代烷,继续反应10-12h。
12、优选地,所述步骤一、二中,萃取后柱层析纯化的具体操作为乙酸乙酯萃取3次,柱层析纯化。
13、第二方面,本发明提供一种α-取代手性膦酸酯类化合物,采用第一方面所述方法合成。
14、说明书附图
15、图1为现有不对称合成方法技术路径图;
16、图2为本发明所述方法的不对称合成方法技术路径图。
技术特征:1.一种α-取代手性膦酸酯类化合物的合成方法,其特征在于,所述合成方法反应式如下式i:
2.根据权利要求1所述的α-取代手性膦酸酯类化合物的合成方法,其特征在于,所述樟脑二醇、三氯化磷、三乙胺、格氏试剂、过氧化氢、lihmds、碘代烷的摩尔比为1:1.1:2.5:1.1:1.1:1.5:1.5。
3.根据权利要求1所述的α-取代手性膦酸酯类化合物的合成方法,其特征在于,所述步骤一、二中,用惰性气体置换的具体操作为用氮气置换三次。
4.根据权利要求1所述的α-取代手性膦酸酯类化合物的合成方法,其特征在于,所述步骤一中,在0℃加入三乙胺和三氯化磷,搅拌10-20min;加入格氏试剂至原料反应完毕;加入过氧化氢氧化,继续搅拌30min。
5.根据权利要求1所述的α-取代手性膦酸酯类化合物的合成方法,其特征在于,所述步骤二中,在-78℃加入lihmds,搅拌30min,然后加入碘代烷,继续反应10-12h。
6.根据权利要求1所述的α-取代手性膦酸酯类化合物的合成方法,其特征在于,所述步骤一、二中,萃取后柱层析纯化的具体操作为乙酸乙酯萃取3次,柱层析纯化。
7.一种α-取代手性膦酸酯类化合物,采用权利要求1~6中任意一项所述方法合成。
技术总结本发明属于化学合成技术领域,具体涉及一种α‑取代手性膦酸酯类化合物的合成方法。本方法首先基于樟脑二醇(CAMDOL)手性辅基获得相应的膦酸酯底物,然后通过LiHMDS对去质子化及碘代烷亲核取代反应,精准构建磷原子邻近叔碳手性中心,进而以高达92%的产率和99:1的非对映选择性,获得结构多样的α‑不对称烷基/芳基化膦酸酯产物,其中包括药物萘普生和布洛芬的磷似物。本发明所述合成方法,底物易得,立体选择性高,产物收率较高,脱除辅基后处理简便。技术研发人员:时恩学,黄煜,张玉龙受保护的技术使用者:中国人民解放军军事科学院防化研究院技术研发日:技术公布日:2024/6/18本文地址:https://www.jishuxx.com/zhuanli/20240619/1667.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表