药物治疗疾病的关键通路预测模型的构建方法及其预测方法
- 国知局
- 2024-09-11 14:25:44
本发明涉及医药及人工智能,更具体地说涉及一种药物治疗疾病的关键通路预测模型的构建方法及其预测方法。
背景技术:
1、高通量“组学”技术的应用促进了人们对药物治疗疾病内在机制的了解。通过生物信息学技术,利用大量基因列表进行功能分析(如基因本体(gene ontology,go)、京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes,kegg))以破解药物治疗疾病的分子机制已成为一种标准做法。
2、功能富集分析是一种应用非常广泛的生物信息学方法,其中kegg通路富集分析应用最为广泛。经典的功能富集分析方法可以识别大量的信号通路,但并非所有的信号通路都与药物治疗疾病过程密切相关。因此,鉴别药物与疾病之间的关键生物信号通路仍然是一个既费钱又费时的研究挑战。
3、目前,预测药物治疗疾病的关键通路通常采用基于统计的富集分析方法。例如,gsea算法能够针对有序的基因集,检测基因表达数据集中显著上调或下调的生物通路。此外,基于网络注释的方法,例如david和kegg数据库,通过超几何分布的富集检验,通常以p值小于0.01和假发现率(false discovery rate,fdr)小于0.05为筛选标准,从无序基因列表中筛选关键通路。然而,基于统计显著性的方法在预测通路时存在局限,它们倾向于识别包含大量基因或基因占比较高的通路,这可能导致通路之间存在较高的冗余度。这种现象与我们精确剖析药物治疗疾病机制的目标并不相符,因为理想情况下,我们希望识别的是那些与疾病治疗直接相关且具有特异性的关键通路。近年来,人们尝试开发新型算法来突出关键的、非冗余的信号通路,以提高预测的关键通路的质量。gomcl使用马尔科夫聚类算法,根据生物学术语之间基因成员的重叠性,将大量术语浓缩成代表非重叠功能主题的聚类,从而预测每个聚类对应的通路作为关键通路。activepathways利用统计数据融合在多个数据集中发现显著富集的通路(即关键通路),使贡献证据合理化并突出显示相关基因。metaboanalyst利用拓扑分析,根据基因在通路中的位置计算通路影响,结合中心性和通路富集结果,评估通路的潜在重要性。此外,还开发了一些基于网络的基因组富集结果可视化方法,如metascape和cytoscape,以加强输出可视化报告并促进数据理解。不过,这些算法都只关注基因集,而忽略了药物与疾病之间的串联关系。
4、总之,目前识别药物治疗疾病的关键通路的方法通常依赖于统计学上的显著性p值和专家的主观判断。这种方法虽然有助于筛选出潜在的重要通路,但存在一些不足之处。它可能导致对那些在人体中广泛影响的信号通路(即那些与多种疾病都有可能相关联的通路)的研究过度集中,而这些通路实际上可能并不是治疗特定疾病的关键。此外,这种方法在富集那些对药物治疗疾病具有特异性的通路方面存在困难,这可能会阻碍对疾病治疗机制的深入理解。
技术实现思路
1、为了克服上述现有技术中存在的缺陷和不足,本发明提供了一种药物治疗疾病的关键通路预测模型的构建方法及其预测方法,本发明的发明目的在于解决上述现有技术单独将各生物实体进行分析,没有考虑和捕捉生物实体的关联,难以富集药物治疗疾病的特异性通路的问题。本发明使用关系图卷积网络(relational graph convolutionalnetwork,r-gcn)学习药物、基因、通路和疾病之间的复杂性生物学关系,并捕捉生物实体之间的相互关系,利用掩码学习,可以根据输入的药物和疾病,计算每条“药物-药物基因-kegg通路-疾病基因-疾病”路径的重要性,从而识别出药物治疗疾病的最重要的通路。本发明更为客观地量化了通路的重要性,且使得通路之间具有可比较性。
2、为了解决上述现有技术中存在的问题,本发明是通过下述技术方案实现的。
3、本发明第一方面提供了一种药物治疗疾病的关键通路预测模型的构建方法,该方法包括以下步骤:
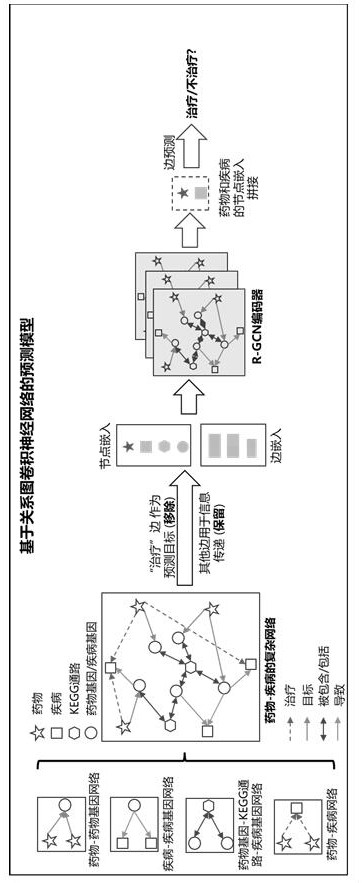
4、s1、数据集的收集步骤:收集“药物-基因”、“基因-kegg通路-基因”、“疾病-基因”和“药物-疾病”的节点和关系数据;
5、s2、图数据的构建步骤:根据s1步骤收集到的数据集构建药物治疗疾病的网络图,该网络图包含5种节点类型和5种边类型,所述5种节点类型分别为药物节点、药物基因节点、疾病节点、疾病基因节点和kegg通路节点,所述5种边类型分别为目标边、被包含边、包含边、导致边和治疗边;药物节点与药物基因节点之间为目标边,药物基因节点与kegg通路节点之间为被包含边,kegg通路节点与疾病基因节点之间为包含边,疾病基因节点与疾病节点之间为包含边,疾病基因节点与疾病节点之间为导致边,药物节点与疾病节点之间为治疗边;
6、s3、边预测模型构建步骤:基于s2步骤构建的药物治疗疾病的网络图,构建一个基于关系图卷积神经网络的边预测模型;在建立边预测模型时,使用r-gcn作为掩码器学习药物治疗疾病的网络图上的节点嵌入,并使用内积计算边的预测概率;将药物治疗疾病的网络图中的治疗边作为预测目标,其他类型的边用于边预测模型的消息传递;
7、s4、解释器构建步骤:在s3步骤构建好的基于关系图卷积神经网络的边预测模型基础上,利用掩码学习思想为该边预测模型构建一个解释器,对每条“药物-药物基因-kegg通路-疾病基因-疾病”路径进行评分并预测关键通路。
8、进一步优选的,所述基于关系图卷积神经网络的边预测模型的构建具体为:
9、初始化s2步骤构建的药物治疗疾病的网络图的表达,将每种类型的节点和边嵌入到同一个特征空间中,假设分别表示节点v和边e的初始嵌入表达,对于每个节点vi∈v,在r-gcn中第l层隐藏层状态被表示为式中,1≤l≤l,l是r-gcn的总层数,表示节点嵌入的权重矩阵,σ(·)表示非线性激活函数,表示节点vi在第l层聚合的邻居信息,表示为式中,r=ψ(e),表示边的类型,ψ(e)表示边类型的映射函数;表示连向vi的邻居集合;表示在第(l-1)层的边类型为r的节点vj的边嵌入;表示沿边类型为r的传递消息的权重矩阵;表示节点vj在第(l-1)层状态;
10、对于治疗边预测任务,给定的药物-疾病对,它们之间缺失治疗边的预测概率表示为式中,表示疾病节点的最终向量表达的转置;表示药物节点的最终向量表达。
11、更进一步优选的,所述基于关系图卷积神经网络的边预测模型的构建过程中,使用交叉熵损失函数优化该边预测模型,所述交叉熵损失函数表示为其中,|vdrug|表示节点类型为药物的总数;|vdisease|表示节点类型为疾病的总数;yij是对之间的黄金标准标签。
12、更进一步优选的,所述非线性激活函数采用leakyrelu激活函数。
13、进一步优选的,s4步骤中,对每条“药物-药物基因-kegg通路-疾病基因-疾病”路径进行评分并预测关键通路,具体是指,使用掩码学习屏蔽某条“药物-药物基因-kegg通路-疾病基因-疾病”路径,基于s3步骤构建的边模型预测计算该条“药物-药物基因-kegg通路-疾病基因-疾病”路径对预测结果的扰动影响,扰动越大,说明该条路径对药物治疗疾病的影响越大,其中的kegg通路越关键。
14、更进一步优选的,假设要掩码的“药物-药物基因-kegg通路-疾病基因-疾病”路径集合为e(m),对于一对药物-疾病对,当没有路径被掩码时,采用s3步骤构建的边模型的预测概率为y,当第i条路径ei被掩码时,基于s3步骤构建的边模型的预测概率其中,表示第i条路径ei被掩码时疾病节点的最终向量表达的转置,表示第i条路径ei被掩码时药物节点的最终向量表达;
15、对于路径ei,它的扰动大小为socre(ei)=|y-y′|;
16、每条路径都经过掩膜计算,获得扰动大小,对所有路径进行排序,路径中越靠前的路径包含的kegg通路则被称为关键通路。
17、进一步优选的,s1步骤中,所述“药物-基因”、“疾病-基因”和“药物-疾病”的节点和关系数据是从治疗靶点数据库中提取出的已批准的药物以及对应的疾病;所述“基因-kegg通路-基因”的节点和关系数据是从kegg通路数据库中提取得到的。
18、更进一步优选的,所述疾病节点统一采用icd-11编码进行规范化编码。
19、本发明第二方面提供了一种基于上述第一方面所述的药物治疗疾病的关键通路预测模型的预测方法,该方法包括以下步骤:
20、a、确定待预测的药物和疾病;
21、b、收集待预测的药物和疾病的“药物-基因”、“基因-kegg通路-基因”、“疾病-基因”和“药物-疾病”的节点和关系数据;
22、c、将b步骤得到的数据集代入到s2步骤得到的药物治疗疾病的网络图中;并利用s3步骤构建的边预测模型计算治疗边的预测概率;然后利用s4步骤构建的解释器对每条“药物-药物基因-kegg通路-疾病基因-疾病”路径进行评分并预测关键通路。
23、与现有技术相比,本发明所带来的有益的技术效果表现在:
24、1、本发明提出了一种基于深度学习的药物治疗疾病的关键通路识别框架,该框架整合了深度学习技术,以识别药物治疗疾病中涉及的关键信号通路。本发明使用关系图卷积网络(r-gcn)来学习药物、基因、通路和疾病之间的复杂生物学关系,并捕捉生物实体之间的相互联系;利用掩码学习,可以根据输入的药物和疾病,计算每条‘药物-药物基因-kegg通路-疾病基因-疾病’路径的重要性,从而识别出药物治疗疾病的最重要的通路。本发明更为客观地量化了通路的重要性,且使得通路之间具有可比较性
25、2、之前的研究单独将各生物实体进行分析,没有考虑和捕捉生物实体的关联。本发明采用关系图卷积网络(r-gcn)来学习药物、基因、通路和疾病之间的复杂生物学关系,并捕捉生物实体之间的相互联系。
26、3、相比之前的技术,本发明更为客观地量化了通路的重要性,且使得通路之间具有可比较性。之前的研究,都是按照基于p值进行通路的筛选,然而p值表示的是一种统计上的显著性,表示基因列表在某个通路上显著富集,而无法代表通路的重要性。
本文地址:https://www.jishuxx.com/zhuanli/20240911/290739.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表