基于图像确定变体致病性的制作方法
- 国知局
- 2024-09-19 14:26:58
本发明所公开的技术涉及人工智能类型计算机和数字数据处理系统以及对应数据处理方法和用于仿真智能的产品(即,基于知识的系统、推断系统和知识采集系统);并且包括用于不确定性推断的系统(例如,模糊逻辑系统)、自适应系统、机器学习系统和人工神经网络。具体地,本发明所公开的技术涉及使用用于转换人工神经网络(ann)或能够通过机器学习来训练的另一类型计算系统的上下文的技术。此外,本发明所公开的技术涉及人工智能型计算机和数字数据处理系统的输入预处理、用于智能仿真的相应数据处理方法和产品,以及实际的预处理输入本身。文献并入以下文献以引用方式并入,即如同在本文完整示出一样,以用于所有目的:2021年10月6日提交的名称为“protein structure-based protein languagemodels”的美国临时专利申请号63/253,122(代理人案卷号illm 1050-1/ip-2164-prv);2021年11月19日提交的名称为“predicting variant pathogenicity fromevolutionary conservation using three-dimensional(3d)protein structurevoxels”的美国临时专利申请号63/281,579(代理人案卷号illm 1060-1/ip-2270-prv);2021年11月19日提交的名称为“combined and transfer learning of avariant pathogenicity predictor using gaped and non-gaped protein samples”的美国临时专利申请号63/281,592(代理人案卷号illm 1061-1/ip-2271-prv);2017年10月16日提交的名称为“training a deep pathogenicity classifierusing large-scale benign training data”的美国专利申请号62/573,144(代理人案卷号illm 1000-1/ip-1611-prv);2017年10月16日提交的名称为“pathogenicity classifier based on deepconvolutional neural networks(cnns)”的美国专利申请号62/573,149(代理人案卷号illm 1000-2/ip-1612-prv);2017年10月16日提交的名称为“deep semi-supervised learning thatgenerates large-scale pathogenic training data”的美国专利申请号62/573,153(代理人案卷号illm 1000-3/ip-1613-prv);2017年11月7日提交的名称为“pathogenicity classification of genomicdata using deep convolutional neural networks(cnns)”的美国专利申请号62/582,898(代理人案卷号illm 1000-4/ip-1618-prv);2018年10月15日提交的名称为“deep learning-based techniques fortraining deep convolutional neural networks”的美国专利申请号16/160,903(代理人案卷号illm 1000-5/ip-1611-us);2018年10月15日提交的名称为“deep convolutional neural networks forvariant classification”的美国专利申请号16/160,986(代理人案卷号illm 1000-6/ip-1612-us);2018年10月15日提交的名称为“semi-supervised learning for training anensemble of deep convolutional neural networks”的美国专利申请号16/160,968(代理人案卷号illm 1000-7/ip-1613-us);2019年5月8日提交的名称为“deep learning-based techniques for pre-training deep convolutional neural networks”的美国专利申请号16/407,149(代理人案卷号illm 1010-1/ip-1734-us);2021年4月15日提交的名称为“deep convolutional neural networks topredict variant pathogenicity using three-dimensional(3d)protein structures”的美国专利申请号17/232,056(代理人案卷号illm 1037-2/ip-2051-us);2021年4月15日提交的名称为“multi-channel protein voxelization topredict variant pathogenicity using deep convolutional neural networks”的美国专利申请号63/175,495(代理人案卷号illm 1047-1/ip-2142-prv);sundaram,l.等人,predicting the clinical impact of human mutation withdeep neural networks.nat.genet.50,1161-1170(2018);jaganathan,k.等人,predicting splicing from primary sequence with deeplearning.cell 176,535-548(2019);2021年4月16日提交的名称为“efficient voxelization for deep learning”的美国专利申请号63/175,767(代理人案卷号illm 1048-1/ip-2143-prv);以及2021年9月7日提交的名称为“artificial intelligence-based analysis ofprotein three-dimensional(3d)structures”的美国专利申请号17/468,411(代理人案卷号illm 1037-3/ip-2051a-us)的优先权。
背景技术:
1、本部分中讨论的主题不应仅因为在本部分中有提及就被认为是现有技术。类似地,在本部分中提及的或与作为背景技术提供的主题相关联的问题不应被认为先前在现有技术中已被认识到。本部分中的主题仅表示不同的方法,这些方法本身也可对应于受权利要求书保护的技术的具体实施。
2、基因组学在广义上也称为功能基因组学,其目的是通过使用基因组规模的测定(诸如基因组测序、转录组谱分析和蛋白质组学)来表征生物体的每种基因组元件的功能。基因组学作为数据驱动的科学出现,其通过从基因组规模数据的探索中发现新特性而不是通过测试预先设想的模型和假设来运作。基因组学的应用包括发现基因型与表型之间的关联、发现用于患者分层的生物标志物、预测基因功能,以及绘制有生化活性的基因组区域(诸如转录增强子)的图表。
3、基因组学数据太大太复杂,以至于不能仅通过可视化研究成对相关来挖掘。相反,需要分析工具来支持发现未预料到的关系,以导出新的假设和模型,并进行预测。机器学习算法与假设和领域专业知识被硬编码的一些算法不同,被设计成自动检测数据中的模式。因此,机器学习算法适合于数据驱动的科学,尤其适合于基因组学。然而,机器学习算法的性能可能强烈依赖于如何表示数据,也就是说,如何计算每个变量(也称为特征)。例如,为了从荧光显微镜图像中将肿瘤分类为恶性或良性,预处理算法可以检测细胞、识别细胞类型,以及生成针对每种细胞类型的细胞计数列表。
4、机器学习模型可以将估计的细胞计数(是手工特征的实例)作为输入特征来对肿瘤进行分类。核心问题是分类性能严重依赖于这些特征的质量和相关性。例如,相关视觉特征(诸如细胞形态、细胞间的距离或器官内的定位)在细胞计数中没有被捕捉到,对数据的这种不完整表示可能降低分类准确度。
5、深度学习(机器学习的分支学科)通过将特征的计算嵌入到机器学习模型本身中以产生端对端模型来解决这个问题。该成果已经通过开发深度神经网络来实现,这些深度神经网络是包括连续基本运算的机器学习模型,其中连续基本运算通过取在先运算的结果作为输入来计算越来越复杂的特征。深度神经网络能够通过发现高复杂度的相关特征(诸如上述实例中的细胞形态和细胞的空间组织)来提高预测准确性。通过数据爆炸、算法的进步以及计算能力的显著增加,特别是通过使用图形处理单元(gpu),已经能够实现深度神经网络的构建和训练。
6、监督学习的目标是获得将特征取作输入并返回对所谓目标变量的预测的模型。监督学习问题的一个示例是预测内含子是否被剪接掉rna上的(目标)给定特征,诸如典型剪接位点序列是否存在、剪接分支点的位置或内含子长度。训练机器学习模型是指学习其参数,这通常涉及使关于训练数据的损失函数最小化,目的是对不可见数据进行准确预测。
7、对于计算生物学中的许多监督学习问题,输入数据可以表示为具有多个列或特征的表格,每个列或特征包含潜在可用于做出预测的数值数据或分类数据。一些输入数据自然地表示为表格中的特征(诸如温度或时间),而其他输入数据需要首先使用被称为特征提取的过程来变换(诸如将脱氧核糖核酸(dna)序列变换为k-mer计数),以符合表格表示。对于内含子剪接预测问题,典型剪接位点序列是否存在、剪接分支点的位置和内含子长度可以是以表格格式收集的预处理特征。表格数据是多种多样监督机器学习模型的标准,范围从简单的线性模型(诸如逻辑回归)到更灵活的非线性模型(诸如神经网络),以及许多其他模型。
8、逻辑回归是二元分类器,即,预测二元目标变量的监督学习模型。具体地,逻辑回归通过使用s型函数(一类激活函数)计算映射到[0,1]区间的输入特征的加权和,来预测正类的概率。逻辑回归或使用不同激活函数的其他线性分类器的参数是加权和中的权重。当用输入特征的加权和不能很好地区分类别(例如,被剪接掉或未被剪接掉的内含子的类别)时,线性分类器失效。为了提高预测性能,可以通过以新的方式(例如,通过取幂或成对乘积)变换或组合现有特征来手动添加新的输入特征。
9、神经网络使用隐藏层来自动学习这些非线性特征变换。每个隐藏层可以被认为是多个线性模型,其输出由非线性激活函数变换,该非线性激活函数诸如s型函数或更流行的整流线性单位函数(relu)。这些层一起将输入特征组成相关的复模式,这有助于区分两个类的任务。
10、深度神经网络使用许多隐藏层,其中一层在每个神经元接收到来自前一层的所有神经元的输入时,被称为是全连接层。神经网络通常使用随机梯度下降来训练,其中随机梯度下降是适合于在非常大的数据集上训练模型的一种算法。使用现代深度学习框架实现神经网络使得能够使用不同的架构和数据集进行快速原型设计。全连接神经网络可以用于许多基因组学应用,包括从序列特征(诸如存在剪接因子的结合基序或序列保守性)预测针对给定序列剪接的外显子的百分比;将潜在致病遗传变体按重要性排序;以及使用诸如染色质标记、基因表达和进化保守性的特征来预测给定基因组区域中的顺式调控元件。
11、为了进行有效的预测,必须考虑空间数据和纵向数据的局部依赖性。例如,打乱dna序列或图像的像素会严重破坏信息模式。这些局部依赖性设置除表格数据之外的空间或纵向数据,对于表格数据,特征的排序是任意的。考虑将基因组区域分类为由特定转录因子结合或不由特定转录因子结合的问题,其中结合区域被定义为染色质免疫沉淀、随后是测序(chip-seq)数据中的高置信度结合事件。转录因子通过识别序列基序与dna结合。基于序列导出特征的全连接层,诸如序列中的k-mer实例的数量或位置权重矩阵(pwm)匹配,可以用于该任务。由于k-mer或pwm实例频率对于在序列内将基序移位具有稳健性,所以此类模型可以很好地推广到具有位于不同位置的相同基序的序列。然而,它们却不能识别转录因子结合依赖于具有明确定义间隔的多个基序的组合的模式。此外,可能的k-mer数量随着k-mer长度呈指数增加,这对存储和过拟合两方面提出了挑战。
12、卷积层是全连接层的一种特殊形式,其中相同的全连接层被局部地(例如在6bp窗口中)应用于所有序列位置。该方法也可以被视为使用多个pwm来扫描序列,例如,针对转录因子gata1和tal1。通过在不同位置使用相同的模型参数,参数总数急剧减少,并且网络能够检测在训练期间未看到的位置处的基序。每个卷积层通过在每个位置处产生标量值来用几个滤波器对序列进行扫描,该标量值量化滤波器与序列之间的匹配度。如在全连接神经网络中那样,在每一层处应用非线性激活函数(通常为relu)。接下来,应用池化操作,其将激活聚集在整个位置轴上的连续仓中,通常取每个通道的最大激活或平均激活。池化减小了有效序列长度,并使信号变得粗糙。随后的卷积层组成前一层的输出,并且能够检测gata1基序和tal1基序是否存在于某个距离范围内。最后,这些卷积层的输出可以用作全连接神经网络的输入,以执行最终的预测任务。因此,不同类型的神经网络层(例如,全连接层和卷积层)可以在单个神经网络内组合。
13、卷积神经网络(cnn)仅在dna序列基础上就能够预测各种分子表型。应用包括对转录因子结合位点进行分类,以及预测分子表型,诸如染色质特征、dna接触图、dna甲基化、基因表达、翻译效率、rbp结合与微小rna(mirna)目标。卷积神经网络除了从序列预测分子表型之外,还可以应用于传统上由手工生物信息学流水线解决的更多技术任务。例如,卷积神经网络可以预测向导rna的特异性、对chip-seq进行去噪、提高hi-c数据分辨率、从dna序列预测来源实验室,以及检出遗传变体。卷积神经网络也已经用于对基因组中的长程依赖性进行建模。尽管相互作用的调控元件在未折叠的线性dna序列上可能远离彼此定位,但这些元件在实际的3d染色质构象中通常彼此邻近。因此,虽然由线性dna序列对分子表型建模是对染色质的粗略近似,但却可以通过允许长范围依赖性和允许模型隐含地学习3d组织的各方面(诸如启动子-增强子成环)来改进。这通过使用扩张的卷积来实现,其具有高达32kb的感受野。扩张的卷积还允许使用10kb的感受野从序列预测剪接位点,从而使得能够跨越与典型的人内含子一样长的距离来整合遗传序列(参见jaganathan,k.等人,predictingsplicing from primary sequence with deep learning.cell 176,535-548(2019))。
14、不同类型的神经网络可以由它们的参数共享方案来表征。例如,全连接层不具有参数共享,而卷积层通过在其输入的每个位置处应用相同的滤波器来施加平移不变性。递归神经网络(rnn)是用于处理实现不同参数共享方案的顺序数据(诸如dna序列或时间序列)的对卷积神经网络的替代方案。递归神经网络对每个序列元素应用相同的操作。该操作将前一个序列元素和新输入作为存储器的输入。该操作将存储器更新并任选地发出输出,该输出被传递到后续层或被直接用作模型预测结果。由于在每个序列元素处应用相同的模型,递归神经网络对于所处理的序列中的位置索引保持不变。例如,递归神经网络可以检测dna序列中的开放阅读框,而不管在序列中的位置是怎样的。该任务需要识别特定系列的输入,诸如起始密码子之后是框内终止密码子。
15、递归神经网络优于卷积神经网络的主要优势在于,在理论上,它们能够经由存储器通过无限长的序列来携带信息。此外,递归神经网络可以自然地处理长度变化很大的序列,诸如mrna序列。然而,在序列建模任务(例如音频合成和机器翻译)方面,与各种技巧(诸如扩张的卷积)组合的卷积神经网络可以达到与递归神经网络相当、甚至更好的性能。递归神经网络可以聚集卷积神经网络的输出,用于预测单细胞dna甲基化状态、rbp结合、转录因子结合和dna可及性。此外,由于递归神经网络应用顺序操作,所以不能轻易并行化,因此计算速度比卷积神经网络慢得多。
16、虽然每个人都有独特的遗传密码,但是人类遗传密码的大部分是所有人共有的。在一些情况下,人类遗传密码可以包括异常值,称为遗传变体,其在相对小群的人群的个体之中可能是共有的。例如,特定的人蛋白质可以包含特定的氨基酸序列,而该蛋白质的变体可以在其他方面相同的特定序列中有一个氨基酸不同。
17、遗传变体可以具有致病性,从而导致疾病。尽管大多数这样的遗传变体已经通过自然选择从基因组中耗尽,但是识别哪些遗传变体可能具有致病性的能力可以帮助研究人员集中于这些遗传变体以获得对相应疾病及其诊断、治疗或治愈的理解。对数百万个人类遗传变体的临床解释仍不清楚。一些最常见的致病性变体是改变蛋白质氨基酸的单核苷酸错义突变。然而,并非所有的错义突变都具有致病性。
18、可以直接从生物序列预测分子表型的模型可以用作计算机扰动工具来探测遗传变异与表型变异之间的关联,并且已经成为用于数量性状基因座识别和变体优先排序的新方法。这些方法非常重要,因为通过复杂表型的全基因组关联分析识别的大多数变体是非编码的,这使得估计它们对表型的作用和贡献具有挑战性。此外,连锁不平衡导致变体的块被共遗传,这在查明单个因果变体方面产生了困难。因此,可以用作评估此类变体的影响的探询工具的基于序列的深度学习模型提供了一种有前途的方法来发现复杂表型的潜在驱动因素。一个示例包括从两种变体在转录因子结合、染色质可及性或基因表达预测方面之间的差异间接预测非编码单核苷酸变体和短插入或缺失(indel)的影响。另一个示例包括根据序列或根据遗传变体对剪接的定量影响,来预测新剪接位点的产生。
19、应用用于预测变体效应的端对端深度学习方法,从蛋白质序列和序列保守性数据预测错义变体的致病性(参见sundaram,l.等人,predicting the clinical impact ofhuman mutation with deep neural networks.nat.genet.50,1161-1170(2018),本文中称为“primateai”)。primateai使用在已知具有致病性的变体上训练的深度神经网络,其中使用跨物种信息进行数据增强。特别地,primateai使用野生型蛋白质和突变型蛋白质的序列来比较差异,并且使用受过训练的深度神经网络来决定突变的致病性。这种利用蛋白质序列进行致病性预测的方法是有前途的,因为其可以避免圆度问题和对先前知识的过度拟合。然而,与有效训练深度神经网络的数据数量充分相比,clinvar中可用的临床数据数量相对较少。为了克服这种数据匮乏,primateai使用常见的人类变体和灵长类动物变体作为良性数据,而将基于三核苷酸背景的模拟变体用作未标记数据。
20、当直接根据序列比对进行训练时,primateai的性能优于现有方法。primateai直接从由约120,000个人类样品组成的训练数据中学习重要的蛋白质结构域、保守氨基酸位置和序列依赖性。primateai在区分候选发育障碍基因中的良性和致病性从头突变方面,以及在复制clinvar中的先验知识方面,明显胜过其他变体致病性预测工具的性能。这些结果表明primateai是变体分类工具的重要进步,可以减少临床报告对先验知识的依赖。
21、蛋白质生物学的核心是理解结构元件如何产生观察到的功能。蛋白质结构数据过剩使得能够开发计算方法来系统地导出支配结构-功能关系的规则。然而,这些方法的性能在很大程度上取决于对蛋白质结构表示的选择。
22、蛋白质位点是蛋白质结构内的微环境,通过其结构或功能作用来区分。位点可以由三维(3d)位置和该位置周围的其中存在结构或功能的局部邻域来定义。合理蛋白质工程的核心是理解氨基酸的结构排列如何在蛋白质位点内产生功能特征。确定蛋白质内各个氨基酸的结构和功能作用提供了有助于工程化和改变蛋白质功能的信息。识别功能或结构上重要的氨基酸允许集中的工程努力,诸如用于改变靶蛋白功能特性的定点诱变。替代性地,这种知识可以有助于避免会破坏期望功能的工程设计。
23、由于已经确定结构比序列保守得多,所以蛋白质结构数据增加提供了使用数据驱动的方法系统地研究支配结构-功能关系的潜在模式的机会。任何蛋白质计算分析的基本方面都是如何表示蛋白质结构信息。机器学习方法的性能通常更多地取决于对数据表示的选择,而不是所采用的机器学习算法。良好的表示高效地捕获最关键的信息,而差的表示产生没有底层图案的噪声分布。
24、蛋白质结构过剩和最近深度学习算法的成功提供了开发用于自动提取蛋白质结构的任务特异性表示的工具的机会。因此,有机会使用3d蛋白质结构的多通道体素化表示作为深度神经网络的输入来预测变体的致病性。
技术实现思路
1、关于一些具体实施,本文描述了用于将蛋白质结构分类的多视图卷积神经网络(cnn)。另外,关于一些具体实施,本文描述了用于此类多视图cnn的经处理输入。在提供此类具体实施和其他具体实施时,本文所述的系统和方法克服了经由人工智能型计算机和数字数据处理系统以及相应的用于智能仿真的数据处理方法和产品对蛋白质结构进行分类中的一些技术问题。另外,本文所公开的技术提供了具体的技术解决方案以至少克服本文所提及的技术问题,以及本文未描述但本领域技术人员认识到的其他技术问题。
2、关于一些具体实施,本文公开了经由可训练计算系统对蛋白质结构进行分类的计算机化方法和用于预处理可训练计算系统的输入的计算机化方法,以及用于实施这些计算机化方法的技术操作的非暂态计算机可读存储介质。该非暂态计算机可读存储介质在其上有形地存储或在其上有形地编码计算机可读指令,这些计算机可读指令在由一个或多个设备(例如,一个或多个个人计算机或服务器)执行时,使得至少一个处理器执行以下方法:(1)经由可训练计算系统对蛋白质结构进行分类的方法,(2)用于预处理可训练计算系统的输入的方法,或(3)用于执行对蛋白质结构进行分类的方法和用于预处理上述输入的方法的组合的方法。
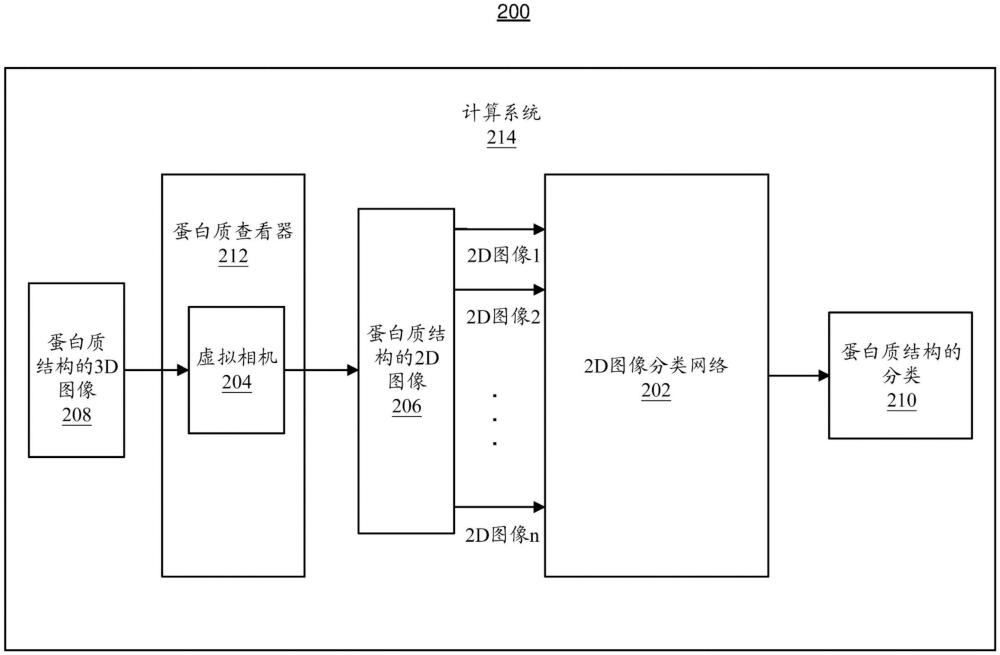
3、在一些具体实施中,确定变体致病性的计算机实现的方法包括:获取蛋白质氨基酸的结构再现;捕获该结构再现的包含来自这些氨基酸的目标氨基酸的那些部分的多个图像;以及至少部分地基于所述多个图像,确定将目标氨基酸突变为另选氨基酸的核苷酸变体的致病性。
本文地址:https://www.jishuxx.com/zhuanli/20240919/298162.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表