一种2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸的制备方法与流程
- 国知局
- 2024-09-14 14:45:40
本发明属于属于精细化工中间体合成领域,涉及一种2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸的制备方法。
背景技术:
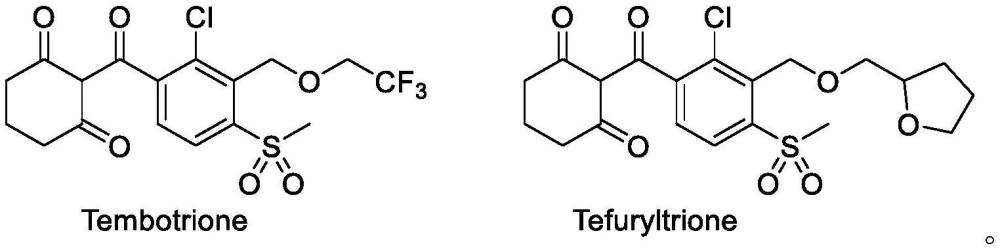
1、对羟基苯基丙酮酸双氧化酶(4-hy-droxyphenylpyruvate dioxygenase,hppd)是目前最重要的除草剂作用靶标之一,hppd抑制剂类除草剂在全球18类除草剂中占有重要地位,销售额占全球除草剂市场的5.8%,此类药剂以广谱、高效、抗性发展缓慢著称。hppd抑制剂类除草剂具有相同作用机理,但结构上不完全相关,有三酮类、吡唑酮类和异噁唑酮类。环磺酮(tembotrione)和呋喃磺草酮(tefuryltrione)是hppd系列三酮类除草剂,结构式如下:
2、
3、环磺酮由拜耳公司于2007年独立开发,环磺酮其活性高于硝磺草酮,对作物安全,主要用于玉米田杂草防治,具有广泛的除草谱;呋喃磺草酮由拜耳、hokko(日本北兴)和zen-noh(日本农协经营的国际贸易公司)联合开发,于2008年在日本上市,呋喃磺草酮主要用于水稻田杂草防治。环磺酮2009年到2014年间的复合年增长率高达51.6%,呋喃磺草酮2009年到2014年间的复合年增长率也高达20.1%,这两个药剂的专利已于2019年9月到期。
4、式ⅰ化合物2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸是此系列化合物的关键中间体,结构式如下:
5、
6、其中,r为三氟乙基时为环磺酮中间体,r为2-四氢呋喃甲基时为呋喃磺草酮中间体。
7、cn112194603a、cn1323292a、us6376429、cn1364160a、cn105601548a、cn1146548c、cn104292137a等均公开了以2-氯-3-甲基-4-甲磺酰基苯乙酮(式ⅱ化合物)为原料,经氧化、酯化、溴化、醚化、水解制备得到式ⅰ化合物,反应式如下:
8、
9、此路线中五步反应总收率在30.0~61.4%之间,总体来看反应步骤长、工艺复杂、设备投资大、收率不高、生产成本相对较高。此外该路线氧化步骤有大量的含盐强酸性废水生成,醚化及水解步骤有大量含氯化钠及溴化钠的强酸性混盐废水生成,处理难度较大。
10、因此,开发一种路线短、工艺简单、收率高、设备投资小、成本低廉、三废易处理的2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸的制备方法,有利于产品的产业化实施,是本领域亟待解决的问题。
技术实现思路
1、针对现有技术的不足,本发明的目的在于提供一种2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸的制备方法。特别是提供一种2-氯-3-(2,2,2-三氟乙氧基)甲基-4-甲磺酰基苯甲酸和2-氯-3-[(rs)-四氢呋喃-2-基甲氧基甲基]-4-甲磺酰基苯甲酸的制备方法。本发明方法工艺路线短、工艺简单、收率高、设备投资小、三废少、三废易处理、成本低廉,很好的克服了目前主流合成工艺所存在的问题。
2、为达此目的,本发明采用以下技术方案:
3、一方面,本发明提供一种2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸的制备方法,所述制备方法包括以下步骤:
4、(1)将2-氯-3-甲基-4-甲磺酰基苯乙酮溶解于溶剂中,使用光源照射下,加入氯化试剂进行氯化反应,得到1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯,反应式如下:
5、
6、(3)1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯在碱性物质存在下与醇r-oh发生缩合反应;或者,1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯与碱金属醇盐rom发生缩合反应,而后酸化,得到2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸,反应式如下:
7、
8、r为三氟乙基或2-四氢呋喃甲基,m表示碱金属。
9、在本发明中,所述原料2-氯-3-甲基-4-甲磺酰基苯乙酮可由现有制备方法制备得到,例如可以由cn105601548a、cn104292137 a、us6376429b1、cn1146548a或cn1323292a中公开的方法制备。
10、在本发明中,提供了2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸的一种全新合成方法,以2-氯-3-甲基-4-甲磺酰基苯乙酮为原料,经氯化、缩合两步反应制备得到目标化合物,目前收率最高可达91.5%,hplc检测含量最高可达98.5%。氯化环节副产4当量的氯化氢,经水吸收后可作为盐酸使用或出售,无废水产生;缩合环节有少量含氯化钠的废水产生,无混合废盐等问题。与原工艺比较,反应步骤由5步缩减到2步,工艺得到大幅简化,且有效减少了设备投资;避免了价格较贵的溴素的使用,且总收率由61.4%提高到91.5%,提高了收率,降低了成本;现工艺只有单一的氯化钠废水生成,与原工艺相比较废水种类少、易处理。
11、优选地,步骤(1)中所述溶剂包括二氯甲烷、氯仿、二氯乙烷或1,1,2-三氯乙烷等卤代烷烃中的任意一种或至少两种的组合,优选为二氯乙烷。
12、优选地,步骤(1)中相对于1mol的2-氯-3-甲基-4-甲磺酰基苯乙酮,所述溶剂的用量为250~1000ml,例如可以是250ml、500ml、750ml或1000ml。
13、优选地,步骤(1)中所述光源为太阳光、高压汞灯、紫外灯、卤素灯和led灯中的任意一种;阳光色温波动较大,一般在1800k-13000k之间,且不具备产业化可行性;高压汞灯色温约4100k、卤素灯色温在2700k-5000k,led灯分为暖白(色温2200k-3500k)、正白(色温4000k-6000k)和冷白(色温约6500k)、紫外灯无色温概念,经试验优选led灯冷白光源,其反应效果最好。
14、优选地,步骤(1)中所述氯化试剂为氯气或磺酰氯中的任意一种,优选为氯气,其性价比更高。
15、优选地,步骤(1)中所述原料与氯化试剂的摩尔比为1:(4.0~5.2),例如可以是1:4.0、1:4.4、1:4.8或1:5.2。
16、优选地,步骤(1)中所述氯化反应的温度为20~80℃,例如可以是20℃、40℃、60℃或80℃。
17、优选地,步骤(1)中所述氯化试剂在5~12h的时间内加入反应体系中,例如可以是5h、8h、10h或12h。在本发明中,所述氯化试剂的加入方式为滴入或通入,当选择的氯化试剂为液体时,采用滴加的方式,当氯化试剂为气体时采用通入的方式。
18、优选地,步骤(1)中所述氯化反应在氯化试剂加入完毕后,保温反应1~4h,例如可以是1h、2h、3h或4h。
19、在本发明中,步骤(1)所述氯化反应中尾气采用两级水吸收得到浓度为30-35%(30%、32%、34%或35%)的盐酸以及浓度5-15%(例如5%、7%、9%、10%、12%或15%)的稀盐酸;其中高浓度的盐酸用于步骤(2)的酸化或者作为副产出售,稀盐酸用于吸收步骤(1)中副产的氯化氢,提浓后用于步骤(2)的酸化中或者作为副产出售。
20、在本发明中,步骤(1)所述氯化反应完成后,进行负压脱溶。
21、优选地,步骤(2)中所述缩合反应的溶剂包括乙腈、thf(四氢呋喃)、dmf(n,n-二甲基甲酰胺)、dma(n,n-二甲基乙酰胺)、nmp(n-甲基吡咯烷酮)、二甲亚砜或二甲砜中的任意一种或至少两种的组合,优选为dmf。
22、优选地,步骤(2)中相对于1mol的2-氯-3-甲基-4-甲磺酰基苯乙酮,所述溶剂的用量为500~1500ml,例如可以是500ml、800ml、1100ml或1500ml。
23、优选地,步骤(2)中所述醇为2,2,2-三氟乙醇或2-四氢呋喃甲醇。
24、优选地,步骤(2)中2-氯-3-甲基-4-甲磺酰基苯乙酮与醇的摩尔比为1:(1.0~1.3),例如可以是1:1.0、1:1.1、1:1.2或1:1.3。
25、优选地,步骤(2)中所述碱性物质为氢氧化钠、氢氧化钾、叔丁醇钠、叔丁醇钾、氢化钠或氢化钾中的任意一种或至少两种的组合。
26、优选地,所述氢氧化钠使用时先制成氢氧化钠水溶液,氢氧化钠的质量浓度为30~96%,例如可以是30%、40%、50%、60%、70%、85%或96%。
27、优选地,所述氢氧化钾使用时先制成氢氧化钾水溶液,氢氧化钾的质量浓度为30~96%,例如可以是30%、40%、50%、60%、70%、85%或96%。
28、优选地,步骤(2)中所述2-氯-3-甲基-4-甲磺酰基苯乙酮与碱性物质的摩尔比为1:(2.0~2.8),例如可以是1:2.0、1:2.2、1:2.4、1:2.6或1:2.8。
29、优选地,步骤(2)中所述碱金属醇盐为2,2,2-三氟乙醇钠、2,2,2-三氟乙醇钾、2-四氢呋喃甲醇钠或2-四氢呋喃甲醇钾。
30、优选地,步骤(2)中所述2-氯-3-甲基-4-甲磺酰基苯乙酮与碱金属醇盐的摩尔比为1:(2.0~2.8),例如可以是1:2.0、1:2.2、1:2.4、1:2.6或1:2.8。
31、优选地,骤(2)中所述缩合反应的温度为-5~15℃,例如可以是-5℃、0℃、5℃、10℃或15℃。
32、在本发明中,步骤(2)所述缩合反应的加料方式并非唯一,将溶剂以及其他反应物(例如碱性物质和醇或碱金属醇盐)置于反应装置中(例如反应釜),滴入1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯进行反应也可;或将溶剂、1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯置于反应装置中(例如反应釜),而后加入其他反应物(例如碱性物质和醇或碱金属醇盐)也可。
33、优选地,步骤(2)中所述碱性物质或碱金属醇盐或1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯在1-5h的时间内加入反应体系中,例如可以是1h、2h、3h、4h或5h。
34、优选地,步骤(2)中所述缩合反应在反应原料加入完毕后,保温反应1~3h,例如可以是1h、2h或3h。
35、优选地,步骤(2)所述酸化为酸化至体系ph值为2以下。
36、优选地,步骤(2)所述酸化使用的酸为盐酸。
37、在本发明中,步骤(2)所述缩合反应结束后,还包括在酸化前进行脱溶的步骤以及在酸化后进行结晶的步骤。
38、作为本发明优选的技术方案,所述制备方法包括步骤如下:
39、(1)将2-氯-3-甲基-4-甲磺酰基苯乙酮溶解于溶剂中,控制温度20~80℃下在5~12h内加入氯化试剂进行氯化反应,经保温反应1~4h后经负压脱溶得到1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯中间体;相对于1mol所述2-氯-3-甲基-4-甲磺酰基苯乙酮,所述溶剂的用量为250~1000ml,所述2-氯-3-甲基-4-甲磺酰基苯乙酮与氯化试剂的摩尔比为1:(4.0~5.2);
40、(2)1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯在碱性物质存在下与醇在-5~15℃下,保温反应1~3h,或者1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯与碱金属醇盐在-5~15℃下发生缩合反应1~3h,后经脱溶、酸化、结晶制备得到2-氯-3-烷氧基甲基-4-甲磺酰基苯甲酸,相对于1mol 2-氯-3-甲基-4-甲磺酰基苯乙酮,所述溶剂的用量为500~1500ml,所述1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯与醇的摩尔比为1:(1.0~1.3),所述1-氯甲基-2-甲磺酰基-6-三氯乙酰基氯苯与碱性物质或碱金属醇盐的摩尔比为1:(2.0~2.8)。
41、本发明所述的数值范围不仅包括上述例举的点值,还包括没有列举出的上述数值范围之间的任意的点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
42、相对于现有技术,本发明具有以下有益效果:
43、本发明提供的制备方法是全新合成方法,以2-氯-3-甲基-4-甲磺酰基苯乙酮为原料,经氯化、缩合两步反应制备得到目标化合物,收率为79%以上,收率最高可达91.5%,hplc检测含量为97%以上,含量最高可达98.5%;氯化环节副产4当量的氯化氢,经水吸收后可作为盐酸使用或出售,无废水产生;缩合环节有少量含氯化钠的废水产生,无混合废盐等问题。
44、与现有技术相比较,本发明整体工艺路线新颖、路线短、条件温和、设备投资小、收率高、三废少、三废易处理、成本低廉,很好的克服了目前主流合成工艺所存在的问题。
本文地址:https://www.jishuxx.com/zhuanli/20240914/295831.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。