一种克服阿霉素耐药的脂质体前药的制备和应用
- 国知局
- 2024-11-06 14:44:38
本发明属于医药技术与药物制剂领域,主要包括克服阿霉素耐药的前药化合物(bqr-ss-dox)的合成和bqr-ss-dox/dspe-peg2000-fa脂质体纳米靶向系统的构建,以及该系统在抗肿瘤中的应用。
背景技术:
1、阿霉素(dox)是临床上广泛应用的化疗药物之一,但肿瘤患者易产生耐药性,一旦出现化疗耐药,肿瘤会迅速发展,这已成为阿霉素治疗肿瘤的主要障碍。因此,迫切需要确定新的治疗方案以克服阿霉素耐药。布喹那(bqr)能够阻断肿瘤细胞的dna修复,可使化疗时dna损伤效应更敏感,同时诱导铁死亡,能够提高化疗药物的抗癌活性。
2、本发明合成构建了一种具有肿瘤靶向作用的bqr-ss-dox/dspe-peg2000-fa还原刺激响应型脂质体纳米递送系统。dspe-peg2000-fa包裹的前药纳米可主动靶向肿瘤细胞表面高表达的叶酸受体;二硫键可选择性地被肿瘤细胞中高浓度gsh降解断裂,释放出布喹那和阿霉素;布喹那可促进肿瘤细胞的铁死亡,阻断肿瘤细胞的dna修复,与阿霉素协同杀伤肿瘤细胞。
技术实现思路
1、本发明的目的是合成一种具有肿瘤靶向性的前药化合物,并将其组装成纳米药物,从而得到稳定性好,具有缓释功能且安全性高的前药脂质体,进而提高抗肿瘤活性和克服阿霉素耐药。
2、本发明通过以下技术方案实现上述目的:
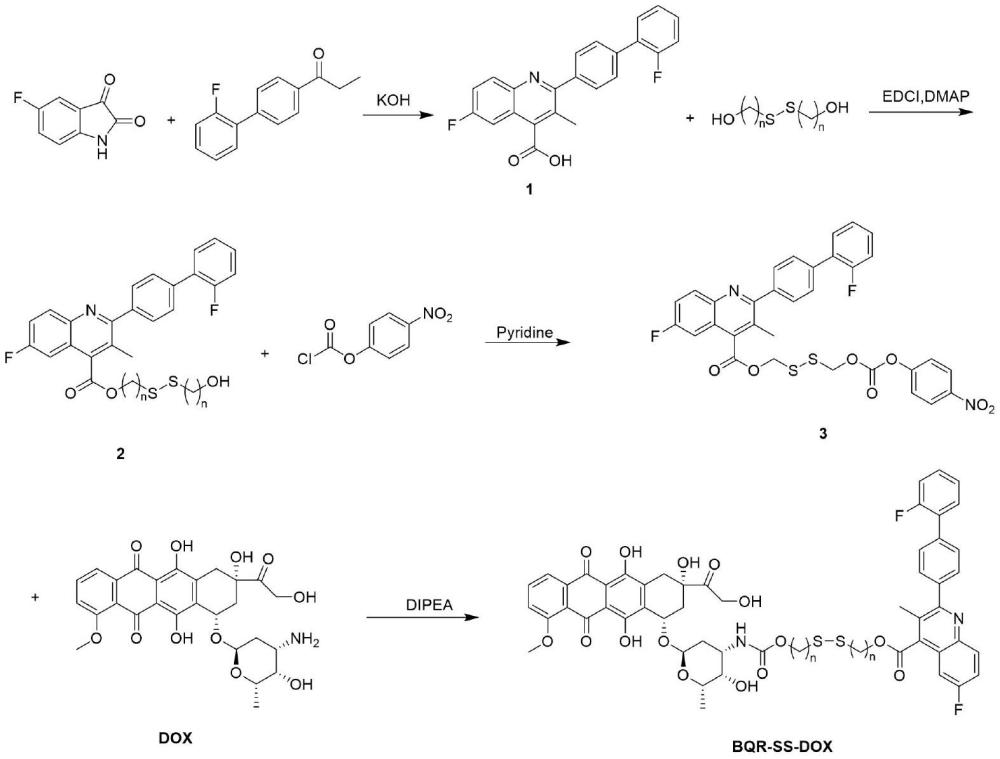
3、本发明所述的前药化合物是通过二硫键将布喹那和阿霉素相连,其结构式如下:
4、
5、其中n为1-6的整数。
6、本发明提供的前药化合物合成方法步骤如下:5-氟靛红和4-(2-氟苯基)苯丙酮为起始原料合成药物布喹那(bqr),使2-羟乙基二硫化物一端的羟基与布喹那相连,另一端的羟基与具有酰氯结构的对硝基苯基氯甲酸酯发生反应并最终与阿霉素相连,得到终产物前药bqr-ss-dox。
7、具体地,本发明提供了前药化合物bqr-ss-dox的合成方法:
8、(1)布喹那的合成:将5-氟靛红溶于无水乙醇/水的混合液中,在室温下加入少量氢氧化钾,搅拌后加入4-(2-氟苯基)苯丙酮,加热回流,冷却至室温,减压蒸干后,加水并用盐酸调ph至2-3,过滤并用乙醚清洗,所得固体为布喹那。
9、(2)6-氟-2-(2'-氟-联苯-4-基)-3-甲基喹啉-4-羧酸2-(2-羟基-乙基二硫基)-乙酯(2)的合成:将布喹那溶于二氯甲烷中,加入edci和dmap室温搅拌后加入2-羟乙基二硫化物,在n2保护下室温反应12h,用水洗涤,将有机层用无水硫酸钠干燥,并减压浓缩。硅胶柱层析法分离得化合物(2)。
10、(3)6-氟-2-(2'-氟-联苯-4-基)-3-甲基喹啉-4-羧酸2-[2-(4-硝基-苯氧基羰基氧基)-乙基二硫基]-乙酯(3)的合成:将化合物(2)溶于二氯甲烷,冰浴下加入吡啶、对硝基苯基氯甲酸酯,室温反应12h。用饱和食盐水洗涤,将有机层用无水硫酸钠干燥,并减压浓缩。硅胶柱层析法分离得化合物(3)。所述含有二硫键的烷基二醇为二硫二甲醇、2,2’-二硫二乙醇、3,3’-二硫二丙醇或4,4’-二硫二丁醇中的一种或多种。
11、(4)6-氟-2-(2'-氟-联苯-4-基)-3-甲基喹啉-4-羧酸2-(2-{3-羟基-2-甲基-6-[3,5,12-三羟基-3-(2-羟基-乙酰基)-10-甲氧基-6,11-二氧-1,2,3,4,6,11-六氢-萘-1-基氧基]-四氢-吡喃-4-基氨基甲酰氧基}-乙基二硫烷基)-乙酯(4)的合成
12、将化合物(3)和盐酸阿霉素溶于dmf中,加入dipea室温反应后加水析出沉淀,过滤得沉淀物,干燥后溶于甲醇:二氯甲烷=1:20的溶剂中,硅胶柱层析法分离得前药化合物bqr-ss-dox。
13、本发明还提供了dspe-peg2000-fa包被bqr-ss-dox前药化合物形成脂质体纳米药物的方法。
14、bqr-ss-dox/dspe-peg2000-fa脂质体纳米的制备方法:本发明采用薄膜分散法进行脂质体的制备;将蛋黄卵磷脂、胆固醇、dspe-peg2000、dspe-peg2000-fa及bqr-ss-dox置于茄形烧瓶内,溶于有机溶剂,40℃下,减压除去有机溶剂,并在瓶壁上成膜。再加入pbs溶液,50℃下水化后,冰水中超声,分别过微孔滤膜(0.45μm、0.22μm、0.1μm)过滤。所述有机溶剂包括乙醇、甲醇、四氢呋喃、二甲基亚砜(dmso)或n,n-二甲基甲酰胺(dmf)中的一种或多种。
15、本发明合成构建了一种具有肿瘤靶向作用的bqr-ss-dox/dspe-peg2000-fa脂质体纳米递送系统。dspe-peg2000-fa包裹的前药纳米可主动靶向肿瘤表面的叶酸受体;二硫键可选择性地被肿瘤细胞中高浓度gsh降解断裂,释放出布喹那和阿霉素;布喹那可促进肿瘤细胞的铁死亡,阻断肿瘤细胞的dna修复,与阿霉素协同杀伤肿瘤细胞。本发明实现了化疗药物对肿瘤细胞的特异性作用,其抗肿瘤效果明显优于抗癌药物的单独使用,具有光明的开发前景。
16、本发明所述的以dspe-peg2000-fa脂质体包载前药化合物构成的bqr-ss-dox/dspe-peg2000-fa脂质体靶向给药系统,其优势在于:(1)脂质体稳定性好,药物包封率高,具有缓释作用,处方工艺可靠;(2)在肿瘤细胞高gsh的环境中,前药中的二硫键与gsh反应发生断裂,释放出活性药物;(3)具有肿瘤靶向性,选择性作用使药物对肿瘤细胞的杀伤效率更高,同时一定程度上避免了抗肿瘤药物损害正常细胞带来的毒副作用,药物协同显著提高了抗肿瘤效果。
17、本发明具有以下效果:(1)设计合成了bqr-ss-dox前药化合物,合成方法稳定,方便可行;(2)制备了dspe-peg2000-fa脂质体包载前药化合物构成的bqr-ss-dox/dspe-peg2000-fa脂质体靶向给药系统,制备方法简单易行,包封率高;(3)实现了两种药物的协同应用及其靶向作用,使抗肿瘤效果得到了显著提升,具有广阔的应用前景。
技术特征:1.一种基于二硫键连接的布喹那(bqr)和阿霉素(dox)的前药化合物,其特征在于,具有如下所示的结构式:
2.权利要求1所述的基于二硫键连接的布喹那(bqr)和阿霉素(dox)的前药化合物的制备方法,其特征在于,包括如下步骤:
3.一种脂质体前药纳米,其特征在于叶酸(fa)与二硬磷脂酰乙醇胺-聚乙二醇(dspe-peg2000)构建成的靶向脂质体(dspe-peg2000-fa)包载权利要求1所述的基于二硫键连接的布喹那和阿霉素的前药化合物组装形成的纳米颗粒。
4.如权利要求3所述的脂质体前药纳米的制备方法,其特征在于,包括如下步骤:将蛋黄卵磷脂、胆固醇、dspe-peg2000、dspe-peg2000-fa及药物bqr-ss-dox置于茄形烧瓶内,溶于有机溶剂后40℃下,减压除去有机溶剂,并在瓶壁上成膜;再加入pbs溶液,50℃水化后,冰水中超声,分别过微孔滤膜(0.45μm、0.22μm、0.1μm)各3遍,制备得到bqr-ss-dox/dspe-peg2000-fa靶向脂质体;所述有机溶剂包括乙醇、甲醇、四氢呋喃、二甲基亚砜(dmso)或n,n-二甲基甲酰胺(dmf)中的一种或多种。
5.如权利要求1所述基于二硫键连接的布喹那和阿霉素的前药化合物或权利要求3所述脂质体前药纳米在制备抗肿瘤药物中的应用。
6.如权利要求1所述基于二硫键连接的布喹那和阿霉素的前药化合物或权利要求3所述脂质体前药纳米在制备注射给药、口服给药或局部给药系统中的应用。
技术总结本发明属于医药物技术领域,涉及一种克服阿霉素耐药的脂质体前药的制备和应用,本发明设计合成了一种二硫键连接布喹那(BQR)和阿霉素(DOX)的前药。在此基础上将前药用构建的靶向脂质体(DSPE‑PEG<subgt;2000</subgt;‑FA)进行包载制备了叶酸靶向的脂质体前药,制备方法简便,稳定性好,实现了药物的高效包载和递送,能够稳定缓释且安全性高。该脂质体前药可靶向肿瘤表面的叶酸受体,在肿瘤细胞内高水平GSH的作用下,二硫键断裂并释放BQR和DOX。BQR能诱导肿瘤细胞铁死亡,并阻碍其DNA修复,可协同阿霉素杀伤肿瘤细胞同时克服细胞对阿霉素耐药。本发明制备的脂质体前药实现了药物的靶向递送,具有较好的体外抗肿瘤活性同时克服阿霉素耐药,具有广阔的发展前景。技术研发人员:牟旭鹏,路海滨受保护的技术使用者:吉林大学技术研发日:技术公布日:2024/11/4本文地址:https://www.jishuxx.com/zhuanli/20241106/323892.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。