一种基于双重协同机制的蛋白质相互作用预测方法
- 国知局
- 2024-09-11 14:15:21
本发明涉及蛋白质预测,特别涉及一种基于双重协同机制的蛋白质相互作用预测方法。
背景技术:
1、探索蛋白质相互作用对理解通路信号传递、分子功能表达等生物学过程具有重要意义。尽管质谱法等实验技术已被应用于蛋白质相互作用的测定,但由于昂贵、耗时的缺点,其仍然无法弥补蛋白质数据爆炸式增长与有限的相互作用见解之间的差距。因此,这种明显的困境促使研究人员开发计算方法来加速蛋白质相互作用预测。
2、早期的蛋白质相互作用计算方法是基于分子动力学模拟的,其根据模拟环境中呈现的一对蛋白形成的复合体的结合姿态来确定蛋白之间的相互作用。然而,除了模拟所需的大量时间成本和计算资源外,这种基于模拟的方法所需的高精度蛋白质真实结构并不总是可得的,这种对蛋白真实结构上的需求有时可能无法实现。
3、随着以数据为驱动的深度学习技术在自然语言处理、计算机视觉等领域的广泛应用,以蛋白质相互作用预测任务为代表的生命科学领域也从中获得启发,且二十一世纪测序技术发展所导致的蛋白质数据激增也为数据驱动的深度学习技术在蛋白质相互作用预测上的应用奠定了基础。
4、目前,基于深度学习的蛋白质相互作用预测方法可以根据其蛋白质表示形式分为两类,即基于蛋白质一维序列的方法和基于蛋白质二维结构的方法。
5、基于蛋白一维序列的相互作用预测模型将一对要预测是否相互作用的蛋白质表示为两个蛋白序列,并将由多个一维卷积层或者全连接层组成的两组隐藏层分别应用于编码后的两个蛋白序列上,进而从蛋白-蛋白对中自动捕获高阶特征。从最后的隐藏层中获得的这两个蛋白的高阶特征被拼接起来作为相互作用分类器的输入,进而使其输出一个特定的0/1值,用于表示两个蛋白间是否发生相互作用。
6、基于二维结构的蛋白质相互作用预测模型在一维序列方法的基础上,将两个蛋白质表示为两个残基接触图(残基为结点,残基间接触关系为边),从而保留了蛋白质的空间结构信息。在此基础上,由多个图神经网络层构成的两组隐藏层被分别应用于两个蛋白内残基间的特征聚合,以进一步提取蛋白质的结构特征,从而获取到比一维序列方法更高的蛋白质相互作用预测准确率。
7、现有的基于一维序列和基于二维结构的蛋白质相互作用预测模型均是将两个要预测是否相互作用的蛋白质看作两个割裂的实体,并通过两组神经网络层对两个蛋白质分别提取特征,从而忽略了两个蛋白质之间的知识共享(如协作蛋白视角下残基重要性的知识共享),相互作用预测准确率还需进一步提高;此外,现有的相互作用预测模型在训练时只以相互作用任务为导向,从而忽略了跨领域知识为相互作用预测所带来的知识互补。因此,本发明通过引入基于交互注意力的蛋白协同机制和基于同构多任务学习的任务协同机制,使得相互作用预测模型不仅能够实现两个蛋白间的知识共享,还实现了这种共享知识在跨领域任务上的互补,从而进一步提高了蛋白质相互作用预测模型的预测准确率。
技术实现思路
1、探索蛋白质相互作用对理解通路信号传递、分子功能表达等生物学过程具有重要意义。尽管质谱法等实验技术已被应用于蛋白质相互作用的测定,但由于昂贵、耗时的缺点,其仍然无法弥补蛋白质数据爆炸式增长与有限的蛋白质相互作用见解之间的差距。因此,这种明显的困境促使研究人员开发基于深度学习的计算方法来加速蛋白质相互作用预测。
2、蛋白质相互作用预测是指向深度学习模型中输入两个蛋白质,从而使得模型可以快速判断两个蛋白质之间是否具有相互作用,显著降低了传统生化实验的经济和时间成本。针对以往蛋白质相互作用预测模型的不足,本发明所解决的技术问题如下:
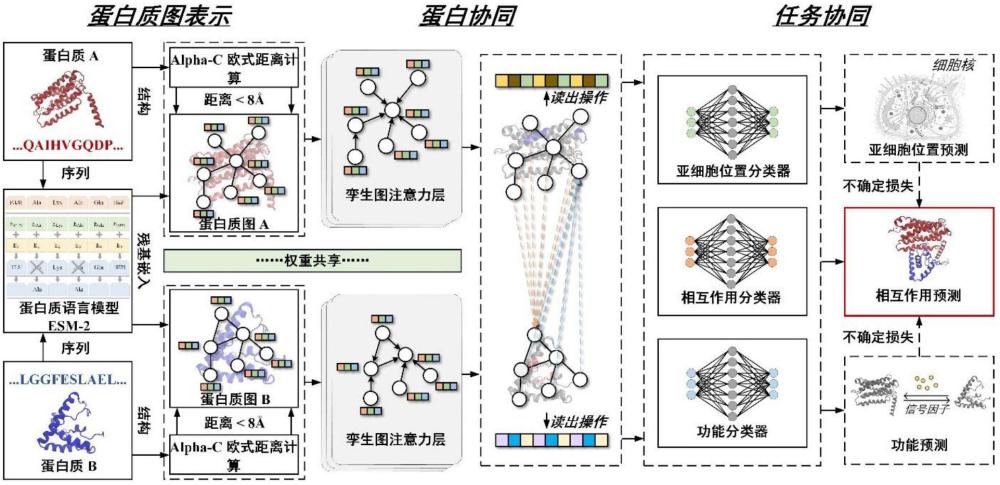
3、(1)本发明从alphafold2所预测的蛋白结构中构建残基接触图,并通过esm-2来计算残基特征,以此来充分构建蛋白质描述符,使得构建的蛋白质描述符包含蛋白空间结构、共进化信息、功能位点等生物规则知识。
4、(2)本发明提出基于交互注意力的蛋白协同机制,使得一个蛋白内关键残基的判定不仅依赖于自身特征,还依赖于其协作蛋白,从而实现了两个蛋白质间的知识共享,进一步提高了模型的相互作用预测准确性。
5、本发明通过收集蛋白质功能和亚细胞位置信息,提出了基于同构多任务学习的任务协同机制,将蛋白质功能预测任务和亚细胞位置预测任务引入到蛋白质相互作用预测模型的训练过程中,从而使得一对蛋白质间共享的知识能够在不同任务中得到互补,进一步提高了模型的相互作用预测准确性。
6、为了弥补现有技术的不足,本发明提供了一种基于蛋白协同和任务协同的蛋白质相互作用预测方法。
7、本发明是通过如下技术方案实现的:一种基于蛋白协同和任务协同的蛋白质相互作用预测方法,包括以下步骤:
8、步骤一:残基接触图的构建
9、首先,根据alphafold2所预测的蛋白结构中alpha-c的三维坐标,计算任意一对残基之间的欧式距离;接着,若残基i和残基j之间的欧氏距离di,j小于预设阈值t,则将其视为接触,依次类推,从而得到完整的接触图,其被定义为:
10、
11、其中,a∈rn×n表示残基接触图,r表示任意实数,n为残基数量,阈值t经实验测定,被设置为此处表示埃米,即10-12米,残基接触图a∈rn×n被看作邻接矩阵,用于表示蛋白质二维结构内残基间的连接关系;
12、步骤二:构建预训练的残基嵌入
13、将长度为n的蛋白序列送入预训练好的esm-2中,通过esm-2将蛋白内的任何残基转化为连续的向量,其尺寸为1×1280;由于esm-2对蛋白共进化信息、功能位点等知识的隐式学习能力,所获得的向量包含了丰富的与蛋白相互作用相关的先验知识,可以直接作为残基嵌入;在此基础上,由堆叠的残基嵌入组成的特征矩阵x∈rn×1280转移到下游的蛋白质任务中;
14、步骤三:利用孪生图注意力层提取蛋白结构特征
15、经过步骤一、二,一对蛋白质可以被表示成两个蛋白质图和其中v(1)和v(2)分别为和的残基结点的集合,a(1)和a(2)分别为和的邻接矩阵,x(1)和x(2)分别为和的特征矩阵;在此基础上,孪生图注意力层被应用,以提取该蛋白质对的结构特征;
16、步骤四、基于交互注意力的蛋白协同
17、经过上述步骤,获得蛋白质对更新后的特征表示,即x(1)′和x(2)′,然而,每种表示的更新只考虑了各自内在的结构或拓扑特征,却忽略了其协同蛋白对自身的信息增强,因此,交互注意力被引入来实现蛋白间的协同,从而促进它们的知识共享;
18、交互注意力的关键是注意力分数的计算,它的计算充分结合了蛋白质对中两个蛋白各自的特征和对此,设计了三个不同的策略用于注意力分数的计算:
19、
20、其中,和分别表示第一个蛋白中的第i个残基嵌入和第二个蛋白中第j个残基嵌入,d表示特征维度,(·)t和⊙分别表示转置和哈达玛积,u∈rd×d,v∈rd×d和w∈r1×d分别是三组可学习的模型参数,用于对和进行可学习的线性映射,从而使得第二个蛋白中第j个残基对第一个蛋白中的第i个残基的注意力分数qij可以被得到;之后进行softmax操作后,获得一对蛋白质的概率向量形式的最终交互注意力分数:
21、
22、
23、其中,exp是以自然常数e为底的指数函数。这样,通过第二个蛋白对第一个蛋白中第i个残基的注意力分数和第一个蛋白对第二个蛋白中第j个残基的注意力分数的计算,蛋白质相互作用的知识在蛋白质对之间被充分共享,使得每个蛋白中对相互作用贡献更大的残基可以根据其协同蛋白被识别出来;最后,将两个注意力得分与原始蛋白质表示和相乘,进行残基重要性的调整,从而实现蛋白质对的协同:
24、
25、
26、步骤五、基于同构多任务学习的任务协同
27、将步骤四得到的蛋白质对的表示s(1)和s(2)直接被输入到全连接分类器中进行二进制相互作用预测,本步骤引入具有蛋白质功能预测和亚细胞位置预测两个辅助任务的同构多任务学习,以共同微调孪生注意力层的可学习参数,从而实现任务协同,促进多个生物领域的知识互补,提高相互作用预测模型的泛化能力。
28、进一步的,步骤三中,孪生图注意力层是两组参数共享的图注意力网络,以蛋白质为例,其残基嵌入被输入到第一组注意力层中,n1为蛋白质的残基数量,图注意力层输出的对应结构特征被表示为:
29、
30、其中,wk∈rd×1280为用于线性映射的可学习参数,li为残基i的一阶邻居残基,其从邻接矩阵a(1)获取,||表示拼接,表示由k头注意力计算的第k个归一化的注意力系数:
31、
32、a∈r2d是可学习权重向量,·t为转置,w∈rd×1280为计算注意力系数的可学习参数,li为残基i的一阶邻居残基;按照这种方式,蛋白质的特征矩阵得以被图注意力层更新,即从而实现进一步的特征提取;
33、此外,对蛋白质的上述操作也被应用于蛋白质由第二组图注意力层对进行结构特征提取,从而被获得,这里第一组和第二组图注意力层参数共享,共同组成孪生图注意力层。
34、进一步的,步骤四中,qij计算过程被归纳成:
35、φ(x(1)′,x(2)′)→q
36、其中,φ:表示策略函数,用于将尺寸分别为n1×d和n2×d的一对特征图映射为尺寸为n1×n2注意力分数矩阵
37、之后,行平均和列平均操作被分别应用于从而得到整个蛋白质对另一蛋白质中单个残基的注意分数和其被定义为:
38、
39、其中,n1和n2分别表示第一个蛋白和第二个蛋白中的残基数量。
40、进一步的,步骤五中具有蛋白质功能预测、亚细胞位置预测辅助任务的同构多任务学习步骤如下:
41、1)蛋白质功能
42、从uniprot数据库中收集了描述蛋白质功能的基因本体术语,用于准确定义每个蛋白质所具有的生物学功能;然后记录出现频率最高的前100个go术语,形成固定的go术语集合;在此基础上,利用词袋编码将蛋白质所具有的功能映射到该固定集合,从而避免了庞大的功能空间所带来的维度爆炸问题;这样,每个蛋白质都可以得到由0和1组成的100维蛋白功能标签yfunc,其中0/1表示一个蛋白质是否具有某个功能。
43、2)亚细胞位置在收集了蛋白质的亚细胞位置信息后,对每个蛋白质的亚细胞位置进行了与蛋白质功能标签相同的预处理操作,因此,还可以获得一个100维的位置标签yloc;此外,亚细胞位置预测和功能预测看作是多标签分类问题,因为一个蛋白质可能在多个亚细胞位置上出现或是具有多种功能;
44、之后,将3组堆叠的全连接层视为各自任务的分类器,用于不同的任务,即蛋白质功能预测、亚细胞位置预测和相互作用预测,定义为:
45、
46、
47、
48、
49、
50、其中,表示模型预测得到的第一个蛋白的功能标签和亚细胞位置标签,表示模型预测得到的第二个蛋白的功能标签和亚细胞位置标签,表示预测得到的两个蛋白质间的相互作用标签,0代表两个蛋白质间没有相互作用,1代表两个蛋白质间有相互作用,[·]表示向量拼接操作,w和b为可学习参数,在两个蛋白质中共享;
51、通过这种方式,蛋白质功能标签、亚细胞位置标签和相互作用的预测标签被得到;
52、在此基础上,根据作为损失函数的二进制交叉熵,三个任务的损失,即和也可以被计算;然而,不同于预定义权重的加权求和或者直接求和,通过不确定损失机制来计算最终损失,从而在早期训练的过程中动态平衡不同任务间的损失尺度,以提高模型泛化性,其被定义为:
53、
54、其中,σ用于量化单个任务的方差和偏移量,它不由人工进行设定,而是被添加到可学习参数集中,从而可以和其它模型参数一起训练。log(1+σ2)表示避免平凡解的正则化项。通过三个任务间的联合训练,孪生图注意力层的可学习参数被三个任务联合优化,从而促进了跨生物领域的知识互补,提高了模型在相互作用预测任务上的预测性能。
55、与现有技术相比,本发明的有益之处为:
56、本发明通过利用蛋白质结构对之间的交互注意力,将蛋白质协同引入到孪生架构中,实现了蛋白质之间的相互作用知识共享。此外,基于同构多任务学习的任务协同被本发明引入,蛋白协同和任务协同相结合,实现了与蛋白质相互作用密切相关的任务之间的知识互补。
本文地址:https://www.jishuxx.com/zhuanli/20240911/289819.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表