一种2-氟-5-甲酰基苯腈的制备方法与流程
- 国知局
- 2024-10-15 09:54:08
本发明属于化学合成,具体涉及一种2-氟-5-甲酰基苯腈的制备方法。
背景技术:
1、2-氟-5-甲酰基苯腈,英文名称2-fluoro-5-formylbenzonitrile,分子式是c8h4fno,白色粉末固体,是一种很好的生物、医药、化学中间体。
2、关于2-氟-5-甲酰基苯腈的合成公开文献报道不多,现有的合成工艺,大体可以分为三类:
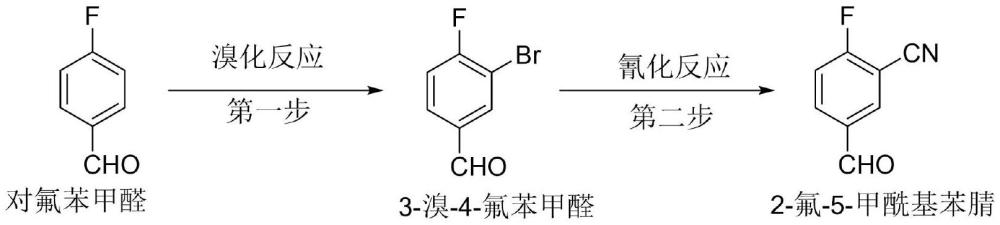
3、方法一、专利文献wo201271684a1,us2013224107a1报导的以对氟苯甲醛为原料,通过溴化、氰化两步反应得到目标产物:
4、
5、第一步溴化反应需要使用液溴或发烟硫酸。液溴属于剧毒、易挥发强腐蚀的原料,发烟硫酸有较强的腐蚀性和危险性;第二步氰化反应需要在高温下进行。全路线对设备和人员防护均有较高的要求,工业化生产受限较多,生产成本高、三废量大。
6、方法二:专利us20026339099b1报导的以2-氟-5甲基苯腈为原料,通过溴化、水解两步反应得到目标产物:
7、
8、第一步溴化反应的溶剂四氯化碳毒性大并且破环臭氧层,使用受到很大限制;反应的选择性较差,收率较低。第二步水解需要使用大量的酸,三废量大、污染高;起始原料的价格也很高,大大增加了物料成本。
9、方法三:专利wo200420414a1报导的以5-溴-2-氟苯甲醛为原料,通过缩合、重排和加成反应得到目标产物:
10、
11、第一步缩合反应使用盐酸羟胺再室温下反应,条件温和,但是起始原料价格昂贵;第二步重排需要使用的流酰氟属于剧毒气体,且对环境危害较大;第三步使用格式试剂反应后与n,n-二甲基甲酰胺加成得目标产物,收率较低。全路线生产成本较高。
技术实现思路
1、本发明的目的是:克服现有技术的不足,提供一种操作简便稳定、产物容易分离、收率高、环境友好、生产成本低、适合工业化规模生产的2-氟-5-甲酰基苯腈的制备方法。
2、本发明合成方法以对氟苯甲醛为原料,经过溴化反应和氰基化取代反应两步反应得到目标产物2-氟-5-甲酰基苯腈;反应方程式如下:
3、
4、本发明所述的2-氟-5-甲酰基苯腈的制备方法,具体步骤如下:
5、1)在第一溶剂中,对氟苯甲醛与溴化试剂发生溴化反应,得到式a化合物;
6、2)在第二溶剂中,所述式a化合物和氰化试剂反应,得到式b化合物,即2-氟-5甲酰基苯腈。
7、步骤1)中,所述第一溶剂为水和酸的混合溶剂,其中酸选自硫酸、盐酸、磷酸等中的一种或多种;优选地,为硫酸(95%~98%)。
8、步骤1)中,所述第一溶剂为水和酸的混合溶剂,其中水和酸的体积比例为4-1:1;优选地,选自4:1、3:1、2:1和1:1中的一种或多种;进一步优选地,为3:1;其中,所述第一溶剂的总体积为原料对氟苯甲醛的体积的8倍。
9、步骤1)中,所述溴化试剂为nbs等。
10、步骤1)中,所述对氟苯甲醛、溴化试剂的摩尔比为1:(1.1-1.5);优选地,该摩尔比为1:1.4。
11、步骤1)中,所述溴化反应的温度为0-40℃;优选地,为20-30℃。
12、步骤1)中,所述酰化反应的时间为6-18h;优选地,为16h。
13、本发明中,步骤1)中得到的产物不进行精制提纯,直接投下一步,提高了收率和降低了成本。
14、步骤2)中,所述第二溶剂为有机溶剂,包括但不限于dmf、dmac、nmp等中的一种或多种;优选地,为nmp。
15、步骤2)中,所述氰化试剂为氰化亚铜等。
16、步骤2)中,所述反应的时间为2-24h;优选地,为12小时。
17、步骤2)中,所述反应的温度为130-160℃;优选地,为130℃。
18、步骤2)中,所述式a化合物、氰化试剂的摩尔比为1:(1-2);优选地,为1:1.5。
19、步骤2)中,还包括后处理步骤;所述后处理中的脱色剂选自活性炭、硅胶等中的一种或多种;优选地,为活性炭。
20、步骤2)中,所述后处理中的脱色剂的使用量为式a化合物投料量的10%wt-30wt;优选地,为10%wt、20%wt和30%wt;进一步优选地,为10%wt。得到颜色较浅的处理液即可。
21、步骤2)中,所述成品的精制选自乙酸乙酯和正庚烷体系、异丙醇、乙醇、异丙醇和正庚烷体系中的一种或多种;优选地,为乙酸乙酯和正庚烷体系。
22、本发明步骤2)采用的氰化试剂为氰化亚铜,避免了超剧毒的氰化钠和氰化钾的使用大大提高了反应的安全性。
23、本发明还提出由所述方法制备得到的2-氟-5-甲酰基苯腈。
24、本发明创新及有益效果包括:在背景技术中方法一的基础上进行大幅度的改进,第一步创造性的采用硫酸和水作溶剂,nbs作为溴源来代替之前的发烟硫酸和液溴的体系,减少了反应的危险性和成本;第二步在130℃左右,少量的高沸点溶剂nmp中,底物和氰化亚铜反应得到最终产物。反应温度比较合适且避免采用剧毒的氰化钠和氰化钾等氰化试剂。本发明一、二两步采用连投的方式,且最终产物的后处理和精制进行了大量优化,降低对环境污染的同时,大大降低了生产成本。此外,本路线的起始原料,关键试剂都是比较廉价易得的,因此物料成本较背景技术中的三种方法都有显著的降低。本发明的综合收率为65%以上,较现有方法整体收率为20~30%,具有实质上的显著提升。综上可见本发明工艺操作简便稳定,产率高、环境友好、综合收率高等优点且原料价廉易得,大幅降低了现有生物、医药、化学中间体的生产成本,具有规模化产业化的广泛应用前景。
25、本发明中所用到的简称对应全称对应表,如下:
26、 entry 缩写 全称 1 nmp n-甲基吡咯烷酮 2 dmf n,n-二甲基甲酰胺 3 dmac n,n-二甲基乙酰胺 4 nbs n-溴代丁二酰亚胺
技术特征:1.一种2-氟-5-甲酰基苯腈的制备方法,其特征在于,所述制备方法,从对氟苯甲醛出发,经过溴化反应和氰基化取代反应两步反应后得到所述2-氟-5-甲酰基苯腈;所述制备方法的反应方程式如下:
2.如权利要求1所述的制备方法,其特征在于,所述制备方法的步骤如下:
3.如权利要求2所述的制备方法,其特征在于,所述步骤1)中,所述第一溶剂为水和酸的混合溶剂,所述水和酸的体积比例为4-1:1;和/或,所述溴化试剂为nbs;和/或,所述反应的温度为0-40℃;和/或,所述反应的时间为6-18h。
4.如权利要求2所述的制备方法,其特征在于,所述步骤1)中,所述对氟苯甲醛、溴化试剂的摩尔比为1:(1.1-1.5)。
5.如权利要求2所述的制备方法,其特征在于,所述步骤1)中得到的产物不进行精制提纯,直接投下一步。
6.如权利要求2所述的制备方法,其特征在于,所述步骤2)中,所述氰化试剂为氰化亚铜;和/或,所述第二溶剂为dmf、dmac、nmp中的一种或多种;和/或,所述反应的温度为130-160℃;和/或,所述反应的时间为2-24h。
7.如权利要求2所述的制备方法,其特征在于,所述步骤2)中,所述式a化合物、氰化试剂的摩尔比为1:(1-2)。
8.如权利要求2所述的制备方法,其特征在于,所述步骤2)中,还包括后处理步骤;所述后处理过程中脱色剂采用活性炭或硅胶中的一种或两种脱色,所述脱色剂使用的量为式a化合物投料量的10%-30%wt。
9.如权利要求2所述的制备方法,其特征在于,所述步骤2)中,成品的精制采用乙酸乙酯和正庚烷体系、异丙醇、乙醇、异丙醇和正庚烷体系中的一种或多种,以此得到纯度大于99%的合格产品,即2-氟-5-甲酰基苯腈。
10.一种如权利要求1-9任一项所述方法制备得到的2-氟-5-甲酰基苯腈。
技术总结本发明公开了一种2‑氟‑5‑甲酰基苯腈的制备方法。本发明制备方法以对氟苯甲醛为原料,经过溴化反应和氰基化取代反应两步反应得到目标产物2‑氟‑5‑甲酰基苯腈。本发明工艺操作简便稳定,每步产物容易分离、产率高、环境友好、综合收率高等优点且原料价廉易得,大幅降低了现有生物、医药、化学中间体的生产成本,有利于工业化规模生产。技术研发人员:杨长圣,张森林受保护的技术使用者:甘肃智资医药有限公司技术研发日:技术公布日:2024/10/10本文地址:https://www.jishuxx.com/zhuanli/20241015/315727.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表