合成阿伐那非、中间体的方法及用途与流程
- 国知局
- 2024-12-06 12:31:36
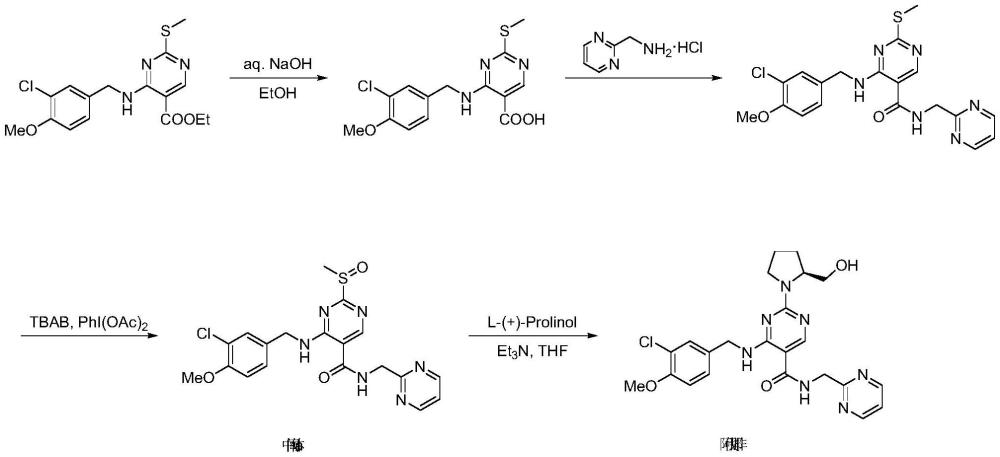
本发明涉及阿伐那非,具体涉及合成阿伐那非、中间体的方法及用途。
背景技术:
1、阿伐那非(avanafil,cas:330784-47-9)是一种口服速效的高选择性磷酸二酯酶-5(pde-5)抑制剂,具有同类药物中起效最快、副作用最小的优势,用于治疗男性勃起功能障碍。
2、目前,阿伐那非的一种通用合成方法如下图1所示。其中的关键步骤是将硫醚氧化得到阿伐那非中间体a(4-[(3-氯-4-甲氧基苄基)氨基]-2-甲亚磺基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺)。该关键步骤的反应选择性优劣及反应生成的杂质高低将极大影响最终制得的阿伐那非纯度和质量控制。因此,开发一种条件温和、高反应选择性、易于后处理纯化的氧化方法,获得高纯度的阿伐那非中间体a,具有极大的工业化价值。
3、目前,阿伐那非中间体a的制备方法主要采用间氯过氧苯甲酸(m-cpba)为氧化剂,国内外专利wo2015177807a1、ep2886540a1、cn103833736a、cn104059025b等都有相关描述。其主要缺点是:
4、(1)间氯过氧苯甲酸的氧化活性较高,尽管投料中多采用滴加但是反应控制依然比较困难,反应选择性较差,硫醚容易被过度氧化到砜(如图7所示,4-[(3-氯-4-甲氧基苄基)氨基]-2-甲磺酰基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺);
5、(2)专利中阿伐那非中间体a未经过纯化直接用于下一步反应,不利于工业大生产中对阿伐那非中间体a的质量控制,最终将影响到生成的阿伐那非原料药的纯度和杂质控制;
6、(3)间氯过氧苯甲酸是管制类化学品,氧化活性比较高,其反应残留如果不及时处理会造成生产危险,在工业化大生产中往往存在安全隐患;
7、(4)副反应多,易生成氮氧化物杂质。这些氮氧化杂质是潜在的基因毒性杂质,且结构与阿伐那非很相似,简单后处理以及重结晶等操作均难以去除该杂质;
8、另外,专利cn103833736a还描述了一种使用次氯酸钠作氧化剂制得阿伐那非中间体a的方法,该方法中使用的是次氯酸钠的固体,而次氯酸钠固体因其稳定性差、易爆的性质,较少用于工业化大生产中,因此该方法的产业化价值不大。
技术实现思路
1、有鉴于此,本发明提供了合成阿伐那非中间体的方法及用途,以至少一定程度上解决上述问题之一。
2、本发明目的之一提供一种合成阿伐那非中间体的方法。所述阿伐那非中间体为4-[(3-氯-4-甲氧基苄基)氨基]-2-甲亚磺基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺。所述方法包括:将含有4-[(3-氯-4-甲氧基苄基)氨基]-2-甲硫基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺和四丁基溴化铵的溶解体系于第一温度条件下混入二醋酸碘苯进行反应。
3、具体地,所述溶解体系中4-[(3-氯-4-甲氧基苄基)氨基]-2-甲硫基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺和四丁基溴化铵的摩尔比为1:(0.02~1)。
4、具体地,所述溶解体系中4-[(3-氯-4-甲氧基苄基)氨基]-2-甲硫基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺和四丁基溴化铵的摩尔比选自1:0.02、1:0.03、1:0.04、1:0.05、1:0.06、1:0.07、1:0.08、1:0.09或1:0.1。
5、具体地,所述溶解体系中的溶剂选自二氯甲烷、乙酸乙酯、甲基叔丁基醚、甲醇、乙醇、乙腈、丙酮、四氢呋喃、n,n-二甲基甲酰胺、n-甲基吡咯烷酮或水中的至少两种。
6、具体地,所述4-[(3-氯-4-甲氧基苄基)氨基]-2-甲硫基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺与混入二醋酸碘苯的摩尔比为1:(1.0~2.0)。例如,所述4-[(3-氯-4-甲氧基苄基)氨基]-2-甲硫基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺与混入二醋酸碘苯的摩尔比选自1:1.01、1:1.02、1:1.03、1:1.04、1:1.05、1:1.06、1:1.07、1:1.08、1:1.09、1:1.1、1:1.11、1:1.12、1:1.13、1:1.14、1:1.15、1:1.16、1:1.17、1:1.18、1:1.19、1:1.20、1:1.21、1:1.22、1:1.23、1:1.24、1:1.25、1:1.26、1:1.27、1:1.28、1:1.29、1:1.30、1:1.31、1:1.32、1:1.33、1:1.34、1:1.35、1:1.36、1:1.37、1:1.38、1:1.39、1:1.40、1:1.41、1:1.42、1:1.43、1:1.44、1:1.45、1:1.46、1:1.47、1:1.48、1:1.49、1:1.50、1:1.51、1:1.52、1:1.53、1:1.54、1:1.55、1:1.56、1:1.57、1:1.58、1:1.59、1:1.60、1:1.61、1:1.62、1:1.63、1:1.64、1:1.65、1:1.66、1:1.67、1:1.68、1:1.69、1:1.70、1:1.71、1:1.72、1:1.73、1:1.74、1:1.75、1:1.76、1:1.77、1:1.78、1:1.79、1:1.80、1:1.81、1:1.82、1:1.83、1:1.84、1:1.85、1:1.86、1:1.87、1:1.88、1:1.89、1:1.90、1:1.91、1:1.92、1:1.93、1:1.94、1:1.95、1:1.96、1:1.97、1:1.98、1:1.99或1:2。
7、具体地,所述第一温度条件选自-10~10℃、-9~9℃、-8~8℃、-7~7℃、-6~6℃、-5~5℃、-4~4℃、-3~3℃、-2~2℃、-1~1℃、-1~10℃、-2~10℃、-3~10℃、-4~102℃、-5~10℃、-6~10℃、-7~10℃、-8~10℃、-9~10℃、-10~0℃、-10~1℃、-10~2℃、-10~3℃、-10~4℃、-10~5℃、-10~6℃、-10~7℃、-10~8℃、-10~9℃、-10℃、-9℃、-8℃、-7℃、-6℃、-5℃、-4℃、-3℃、-2℃、-1℃或0℃。
8、具体地,所述方法还包括:使用亚硫酸钠水溶液对反应进行淬灭的步骤;以及对淬灭的反应物进行纯化得到所述阿伐那非中间体的步骤。
9、具体地,所述亚硫酸钠水溶液的浓度为5%~20%。例如,所述亚硫酸钠水溶液的浓度为5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%或20%。
10、具体地,所述纯化的步骤包括:对淬灭的反应物进行萃取,获得有机相;对所述有机相进行水洗、浓缩至干和重结晶。
11、具体地,在反应的第6~18h进行淬灭。
12、具体地,对淬灭的反应物进行萃取的步骤包括:对所述淬灭的反应物中静置后的水相进行二氯甲烷萃取。
13、具体地,所述重结晶采用丙酮进行。
14、具体地,所述重结晶采用四氢呋喃进行。
15、具体地,对浓缩至干的固体物,使用丙酮,升温(30~60℃)回流0.5h,然而低温(-10~10℃)下析出晶体2h,过滤,烘干。
16、具体地,继续将该蒸干的固体物混入四氢呋喃中,升温(30~70℃)回流0.5h,然后低温(-10~10℃)下析晶2h,过滤,烘干。
17、本发明目的之一还提供了合成阿伐那非的方法。该方法包括:将含有4-[(3-氯-4-甲氧基苄基)氨基]-2-甲硫基-n-(嘧啶-2-基甲基)嘧啶-5-甲酰胺和四丁基溴化铵的溶解体系第一温度条件下混入二醋酸碘苯进行第一反应;对所述第一反应进行淬灭后,所得反应物萃取,得到有机相;向所述有机相中加入三乙胺和l-脯氨酸于第二温度进行第二反应,得到所述阿伐那非。
18、本发明目的之一还提供了合成阿伐那非中间体的方法在制备阿伐那非中间体或阿伐那非中的用途。
19、本发明提供的合成阿伐那非中间体的方法,以四丁基溴化铵作催化剂,催化氧化制备阿伐那非中间体a。由于该反应过程能够受到该四丁基溴化铵的控制,反应无需滴加方式加料,并且该方法提供的反应选择性好,降低了硫醚的过度氧化发生率。
20、另外,该方法提供的反应物仅需简单重结晶纯化,即可得到高纯度的阿伐那非中间体a的白色粉末,进而可以很方便的进行阿伐那非中间体a的质量控制,而这也有利于后续阿伐那非原料药的质量控制及杂质控制。
21、并且,该方法的反应条件温和,操作简便,阿伐那非中间体a收率高,纯度高,具有应用于制备阿伐那非中间体或阿伐那非中的良好工业化前景。
22、此外,该方法采用二醋酸碘苯作为氧化剂,二醋酸碘苯性质稳定,易存放、安全风险低、毒性小,相较于之前专利报道的次氯酸钠(易分解)、间氯过氧苯甲酸(管制类化学品)和双氧水(管制类化学品)等氧化剂,生产安全性和便利性大大提高。
本文地址:https://www.jishuxx.com/zhuanli/20241204/341921.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。
下一篇
返回列表