基因组遗传标记鉴定与跟踪人源样本的方法及应用
- 国知局
- 2024-10-15 09:21:44
本发明涉及细胞生物学和生物信息学领域,具体涉及一种基因组遗传标记鉴定与跟踪人源样本的方法及应用。
背景技术:
1、原核和真核生物的基因组中含有丰富的短串联重复序列(short tandem repeat,str),约占基因组序列的3%,在人类基因组中有超过100万个微卫星位点,也就是str位点。str序列通常以1-6个碱基为单元的核心序列(如:tctatctatcta)首尾相连多次重复形成的短串连序列。研究表明str位点具有高多态性,遗传稳定性,而在不同个体之间相同位置的位点重复单元数可能不同。因而各str构成其核心序列重复数目(序列长度)在不同种族、人群和个体之间具有遗传的差异多态性,可作为基因组高度多态性的标记。str在基因组中的种类非常多,通过检测不同的str位点可以创建特定个体的独特基因档案进而可以对个体进行身份鉴定识别。目前str检测已成为鉴定方法中的一种,已被广泛应用于个体识别、司法鉴定和实验室鉴定等。
2、实验室细胞系交叉污染是常有发生的问题。str分析是鉴定一个细胞系真实性的方法。为了常规细胞系鉴定,根据构建的细胞系str遗传特征谱,建立了各种细胞系str特征谱的数据集库,如美国模式培养物集存库(atcc,search-str-database,即atcc-str9特征谱)、德国菌种保藏中心(dsmz,str profile database–iclac)和日本细胞保藏中心(jcrb)等。近来,人源细胞系的str鉴定法已经建立的相关的国际标准,参照ansi/atcc asn-0002-2022《authentication of human cell lines:standardization of short tandemrepeat(str)profiling》。当时的分析标准方法是采用多重荧光pcr毛细管电泳法(ce法),这也是当前公认的细胞系检测的金标准。
3、ce法检测也有一些技术局限性。第一,传统的ce法需要购买特定的仪器分析仪器和软件并且只能检测str位点的长度信息。这些配置导致atcc-str9特征谱的ce法检测成本昂贵,阻碍其发展为实验室的常规检测方法。第二,传统的ce法只能鉴定str长度(str重复单元数),无法区分str突变造成的str结构变化,所以只能人为忽略1-2个str重复单元。也就是说,ce法测定出来的str长度可能与实际str长度不同。第三,ce法检测无法与特定dna测序一起完成。所述的特定dna包括人类基因组中基因突变、基因工程插入基因组的dna片段或者基因编辑导致的基因变化等。例如,在遗传性疾病和肿瘤等患者的基因组中存在与疾病相关的重要基因突变。随着基因编辑技术的快速发展,在人类细胞的基因组中进行人工改造也成为了疾病治疗的潜在手段,例如car-t细胞治疗技术。针对上述疾病细胞的特定dna突变和工程细胞内的改造dna序列的鉴定也是现代基因工程和基因编辑技术的重大诉求。但ce检测无法同时完成此类dna片段的序列识别。因此同时完成细胞的str鉴定和特定dna鉴定手段是非常重要的。
4、深度测序分析法也可以用于细胞鉴定,而且比ce检测法有不少优势。深度测序分析法与ce法相比,深度测序揭示了str位点的真实变化,也带来更多的变化和更多的技术挑战。而ce法只能通过标准品的对比而获得str长度,从而只能检测预设和有限的str位点;深度测序分析法可以同时检测预设或者定制的更多str位点。此外,深度测序分析法可以利用引物添加标签序列实现多样本的同时检测,从而降低成本。随着深度测序技术的不断突破,高通量,低耗时和低成本的优势将日益突显。另一方面,深度测序分析法来做细胞鉴定也存在一些技术挑战,如在全基因组测序的str分析中,每个靶向的str位点得到的测序深度不够多且一定的杂信号序列;同时str位点存在的个体序列变异也会导致深度测序分析结果不一致。这些技术上的挑战也制约了深度测序分析法在细胞鉴定领域的应用。
5、总之,研发一种既能利用深度测序方法多维度精准分析str序列和长度,兼容原ce法的细胞数据库,并且升级覆盖细胞中特定dna序列(例如基因编辑dna突变和基因工程插入dna等)的人源细胞鉴定与示踪方法是本发明所解决的关键问题。
技术实现思路
1、本发明的术语和声明:
2、本发明中,术语“遗传标记”是指可追踪染色体、染色体某一节段、某个基因位点在家系中传递的任何一种遗传特性。
3、本发明中,术语“测序”是指对dna的序列进行判断的一种方法。
4、本发明中,术语“深度测序”是指通过第二代测序技术高通量获得dna序列的一种方法。
5、本发明中,术语“深度测序read序列”是指通过第二代测序技术判断dna序列而产生的短dna序列信息。
6、本发明中,术语“深度测序读数”是指通过采用第二代测序技术获得dna序列的高通量read数。
7、本发明中,术语“等位基因”是指位于一对同源染色体相同位置上控制同一性状不同形态的基因,本发明的等位基因由相同位点表现出不同str重复单元数所体现。
8、本发明中,术语“杂合率”是指同源染色体在同一基因位点上的两个等位基因不相同出现的概率。
9、本发明中,术语“侧翼序列”是指重复序列两端的序列。
10、本发明中,术语“gc含量”是指鸟嘌呤和胞嘧啶所占的比率。
11、本发明中,术语“染色体”是指细胞在有丝分裂或减数分裂时dna存在的特定形式,由染色质丝螺旋缠绕,逐渐缩短变粗形成。染色体有种属特异性,随生物种类、细胞类型及发育阶段不同,其数量、大小和形态存在差异。
12、本发明中,术语“突变基因”是指基因组dna分子发生的偶然的、可遗传的变异现象。基因突变从分子水平上看是指基因在结构上发生碱基对组成或排列顺序的改变,可以发生在发育的任何时期,通常发生在dna复制时期,即细胞分裂间期。随着基因技术的发展,“突变基因”也包括通过人为改造基因组dna分子发生的变异现象,并可以稳定复制到下一代细胞中。
13、本发明中,术语“碱基”是指嘌呤和嘧啶的衍生物,是核酸、核苷、核苷酸的成分。
14、本发明中,术语“str”是指原核和真核生物的基因组中含有丰富的短串联重复序列(short tandem repeat,str)。
15、本发明中,术语“str标记集”是指一套优选人类基因组的遗传标记集,包括str位点和x/y染色体特征位点。
16、本发明中,术语“str特征谱”是指通过str标记集建立每个人源样品的特征谱,特征谱包括str标记集位点和相对应str重复单元总数。
17、本发明中,术语“stram分析”是指对str序列、x/y染色体特征序列和特定dna序列的在深度测序数据后的自动化与精准化的生物信息分析方法。
18、本发明中,术语“rsfr”是指str位点分析和str侧翼分析的read数比值。
19、本发明中,术语“cstr分析”是指str位点分析和str侧翼分析所得的str长度对比值。
20、本发明中,术语“nru”(number of repeat units)是每个str位点的str碱基长度除以str重复单元碱基数而获得的重复单元总数;术语“nru-s”(number of repeat unitsby str analysis)是指str位点分析所得str长度除以str重复单元的碱基数的数值。术语“nru-f”(number of repeat units by flank analysis)是指str侧翼分析所得str长度除以str重复单元的碱基数的数值。nru需要通过对比nru-s和nru-f核实或者校正而最终产生的每个位点的str重复单元数;同时也是str特征谱中每个str位点的数值。
21、本发明的目的是建立一种用基因组遗传标记鉴定与跟踪人源样本的方法。
22、本发明技术方案包括:
23、第一方面,本发明提供了一种精准鉴定与跟踪人源样本的方法,包括以下组件:
24、(1)一套优选人类基因组的遗传标记集,即str标记集;
25、(2)靶向str标记集的pcr引物组、特定dna序列的pcr引物设计和优化的pcr扩增方法;
26、(3)针对str标记集中各位点序列和特定dna序列扩增产物的深度测序以及针对深度测序数据的自动化生物信息分析,即stram分析;
27、组件(3)中所述的深度测序数据的自动化生物信息分析的方法包括:
28、str位点分析、str侧翼分析、x/y染色体分析和特定dna序列分析;
29、所述的str位点分析和str侧翼分析同时进行,以完成str序列的自动化生物信息分析;
30、所述的str位点分析是将深度测序read序列定位到对应参考人类基因组,并且进一步分析获得str重复单元、str长度信息和定位到特定str位点的测序读数,即read数;
31、所述的str侧翼分析是将深度测序read序列与参考人基因组中str侧翼序列进行比对分析,选出与参考人基因组中str侧翼序列一致的深度测序read,然后测算每一个匹配的read序列中str两侧翼之间的str序列长度;
32、所述的str位点分析和str侧翼分析的结果对比包括在相同str位点上read数对比和str长度对比;
33、所述的x/y染色体分析和特定dna序列分析是将深度测序read序列与参考x/y染色体特征序列和参考特定dna序列进行比对分析,选出含有x/y染色体或者特定dna序列的测序read,并测算含x/y染色体和特定dna匹配序列的read总数。
34、具体的,上述结果校对对比不一致的原因为str序列或侧翼序列至少一种发生突变。
35、具体的,组件(1)所述的str标记集包括:str位点和x/y染色体特征位点。
36、进一步具体的,所述组件(1)str标记集中的位点,覆盖了人类基因组的22对常染色体和xx或xy性染色体,并且每个常染色体至少包括1个str位点,以及x/y性染色体上一个特征位点。而且优选这些位点出来,是为了适配pcr、深度测序和生物信息分析。
37、具体的,上述的str标记集的所有位点优选适用于深度测序。
38、具体的,组件(2)所述的引物组的核苷酸序列如seq id no:1-70所示。
39、具体的,所述的组件(2)中包括设计靶向str标记集位点的引物组。
40、具体的,所述的组件(2)引物组扩增的dna序列包括组件(1)str标记集的所有位点,每个位点均至少有1对引物。
41、可选地,所述的str位点的引物组设计也可扩展其他str位点进行引物设计与鉴定。
42、优选的,所述的组件(2)中pcr产物大小不能超过所选深度测序长度限制,如根据双末端测序(paired-end sequencing,pe)的要求设计不同的pcr产物大小,优选的,pe150的pcr产物大小不超过300bp;pe200的pcr产物大小不超过400bp,且须包含全部的str序列以及5’和3’两端的侧翼序列10-50bp。
43、优选地,所述str位点的引物设计为常规设计或进行引物标签添加设计。
44、进一步优选,所述的引物标签包括但不限于特异片段、文库构建常用序列。
45、具体的,所述的引物标签添加可用于样本的区分与高通量分析,增加检测的样本量同时降低样本检测成本。
46、具体的,所述组件(2)中特定dna序列的引物通用性设计与鉴定是为了对特定的dna序列进行鉴定与追踪。
47、具体的,所述组件(2)中所述特定dna序列的pcr引物设计根据深度测序要求优化产物长度且包含特殊dna序列信息。
48、进一步具体的,特定dna序列引物设计包括突变序列,pe150对应的是不超过300bp的pcr产物;pe200对应不超过400bp的pcr产物。
49、具体的,所述组件(2)中特定dna序列选自特定插入或删除dna片段、肿瘤细胞携带的基因突变、基因编辑的插入或删除dna序列(包括基因片段)中的一种或多种。
50、具体的,组件(2)中基因特定位点突变鉴定为用深度测序和stram分析方法检测特定基因位点的碱基突变情况。
51、具体的,特定序列的引物通用性设计与鉴定包括肿瘤常见基因突变点,基因编辑插入或删除dna序列(包括基因片段)进行引物设计,进而可以实施靶向基因位点的突变鉴定分析和监控检测。
52、具体的,所述组件(2)中靶向鉴定特定dna序列适用于相同来源但不同基因型的细胞鉴定或者基因工程改造的克隆细胞的溯源和鉴定。进一步具体的,所述组件(2)中技术可用于基因突变点的鉴定分析。
53、进一步具体的,所述组件(2)中技术可用于鉴定来源相同但带有不同dna突变的不同样本。
54、具体的,所述的组件(2)中包括str位点的扩增技术。
55、再进一步具体的,所述的str位点的扩增技术中高保真dna聚合酶选自primestardna聚合酶、superfi ii dna聚合酶、phanta max super-fidelity dna聚合酶中的一种或多种,高保真dna聚合酶的选择用于保证测序序列的高保真性和降低str的stutter信号。
56、具体的,将所有str位点的引物采用统一的退火温度;
57、进一步具体的,所述退火温度为50-65℃。
58、具体的,所述的组件(3)中stram分析,还包括str位点分析和str侧翼分析的比对分析和人工校正;所述的str位点分析和str侧翼分析的结果对比和人工校正,包括在相同str位点上read数对比和str长度对比,即通过rsfr参数和cstr参数值进行确定;
59、rsfr参数:指在同一个str位点上通过str位点分析和str侧翼分析所得的read数差异比值(ratio of str read number to flank reads,rsfr)。该参数值的解读:a)rsfr值在(-0.05,0.05)之间,表示该str位点的str位点分析和str侧翼分析正常,不需要校正分析;b)rsfr的值在(-1,-0.05)和(0.05,1)之间,表示str位点分析和str侧翼分析至少一个出现异常,需要校正分析;rsfr的计算公式:
60、
61、cstr参数:指在同一个str位点上通过str位点分析和str侧翼分析所得的str长度比值(comparison of str length from str analysis and str length from flank endanalysis,cstr)。该参数值的解读:a)cstr值是“true”,代表str位点分析和侧翼分析的str长度一致,即该str位点无突变,不需要校正分析;b)cstr值是“false”,代表str位点分析和侧翼分析的str长度不一致,即str序列或侧翼序列出现突变现象,需要进行校正分析。
62、具体的,所述的str位点分析和str侧翼分析的比对分析和人工校正是用于校正str及两端侧翼序列的突变造成的str位点分析和str侧翼分析差异。
63、具体的,校正后得到str位点特征谱各位点对应的真实str重复单元数。进一步具体的,所述数据采用生物信息学软件或在线工具进行分析。
64、再进一步具体的,深度测序数据的stram分析方法中str位点分析和str侧翼分析采用生物信息学软件或网页在线工具包括但不限于r语言、galaxy服务器、str detection、lobstr、repeatseq、hipstr、stretch、gangstr。stram分析方法中x/y染色体分析和特定dna序列分析采用ncbi blast、gapped blast、psi-blast、mummer4、nucmer等。
65、具体的,所述人源样本选自人源组织、细胞、基因组dna、人源细胞制剂、人源细胞系、体外人源原代培养细胞和组织、外源人源基因工程细胞、人源基因编辑细胞、异种移植的人源组织、源自健康人体或者患者的类器官中的一种或多种。
66、再进一步具体的,所述的人源细胞系包括但不限于hek293ft细胞系、a549细胞系、sw480细胞系、sw620细胞系、jurkat e6.1细胞系、hs578t细胞系、a549细胞系和t47d细胞系等。
67、再进一步具体的,所述人源细胞制剂包括但不限于干细胞、免疫细胞和它们分化的细胞。
68、再进一步具体的,所述外源人类基因工程细胞包括但不限于car-t细胞、car-nk细胞。
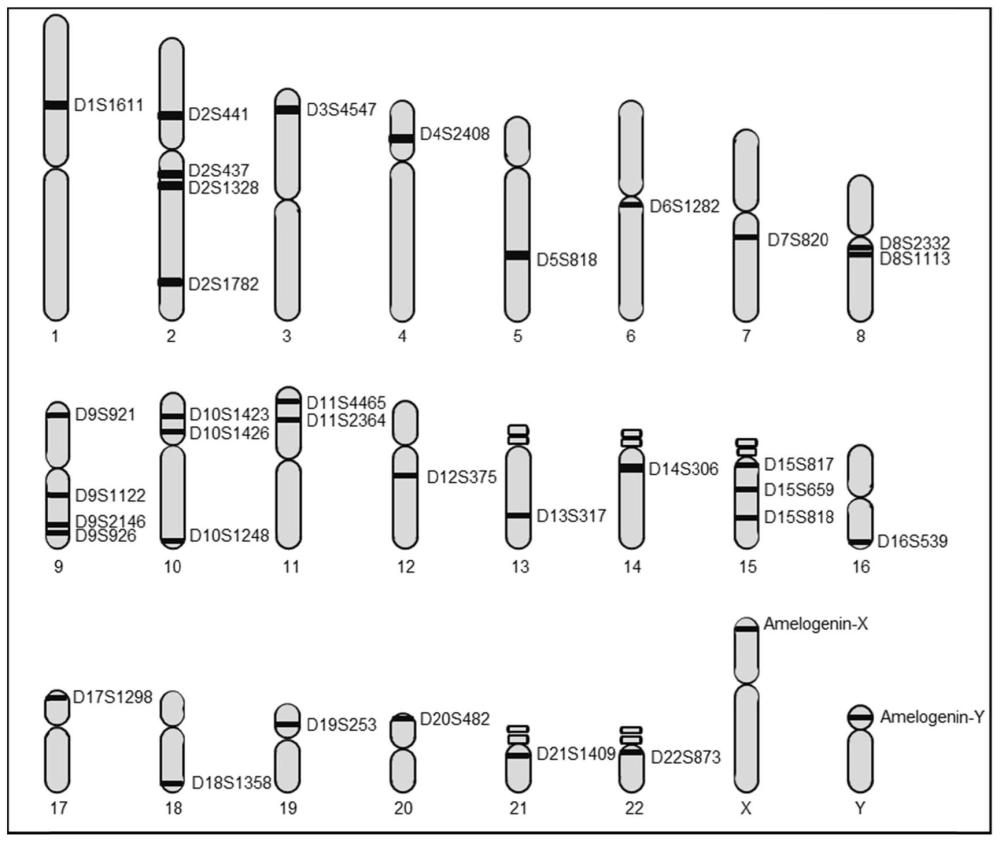
69、又一方面,本发明提供了一套优选人源样本鉴定或溯源的str标记集,所述的str标记集包括:d1s1611、d2s437、d2s1328、d2s1782、d2s441、d3s4547、d4s2408、d5s818、d6s1282、d7s820、d8s1113、d8s2332、d9s921、d9s926、d9s1122、d9s2146、d10s1248、d10s1423、d10s1426、d11s2364、d11s4465、d12s375、d13s317、d14s306、d15s817、d15s659、d15s818、d16s539、d17s1298、d18s1358、d19s253、d20s482、d21s1409、d22s873和amelogenin。
70、又一方面,本发明提供了在上述的str标记集的基础上根据上述方法构建的str特征谱,所述的str特征谱包括从str标记集中挑选靶向每条染色体的一个或多个str位点和stram分析所得的str重复单元数。
71、进一步具体地为str特征谱优化选择的str标记集:d1s1611、d2s1782、d3s4547、d4s2408、d5s818、d6s1282、d7s820、d8s2332、d9s926、d10s1426、d11s2364、d12s375、d13s317、d14s306、d15s659、d16s539、d17s1298、d18s1358、d19s253、d20s482、d21s1409、d22s873和amelogenin。
72、又一方面,本发明提供了上述的方法或str标记集或str特征谱在人源样本的溯源、dna信息追踪、基因编辑dna追踪、插入dna追踪、细胞制剂和细胞系的质量监控中的应用。
73、本发明的有益效果包括:
74、本发明的方法可用于人源细胞的精准str遗传标记鉴定,同时包括特定dna序列(例如基因工程或者基因编辑序列等)鉴定。基于二代深度测序高通量快速地靶向检测基因组中的短串联重复序列,为str序列的检测降低成本和分析程序。可同时采集和鉴定人源细胞的特征str位点数据和基因突变(或者基因改造),从而大大提高人源细胞溯源与鉴定的流程精准度和检测范围。由于该方法具有更精准、更多维度、更低成本的优势,其将在细胞治疗等领域的质量监控中发挥极大的作用。
本文地址:https://www.jishuxx.com/zhuanli/20241015/313898.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。