Cas12a切口酶的制作方法
- 国知局
- 2024-10-21 14:19:19
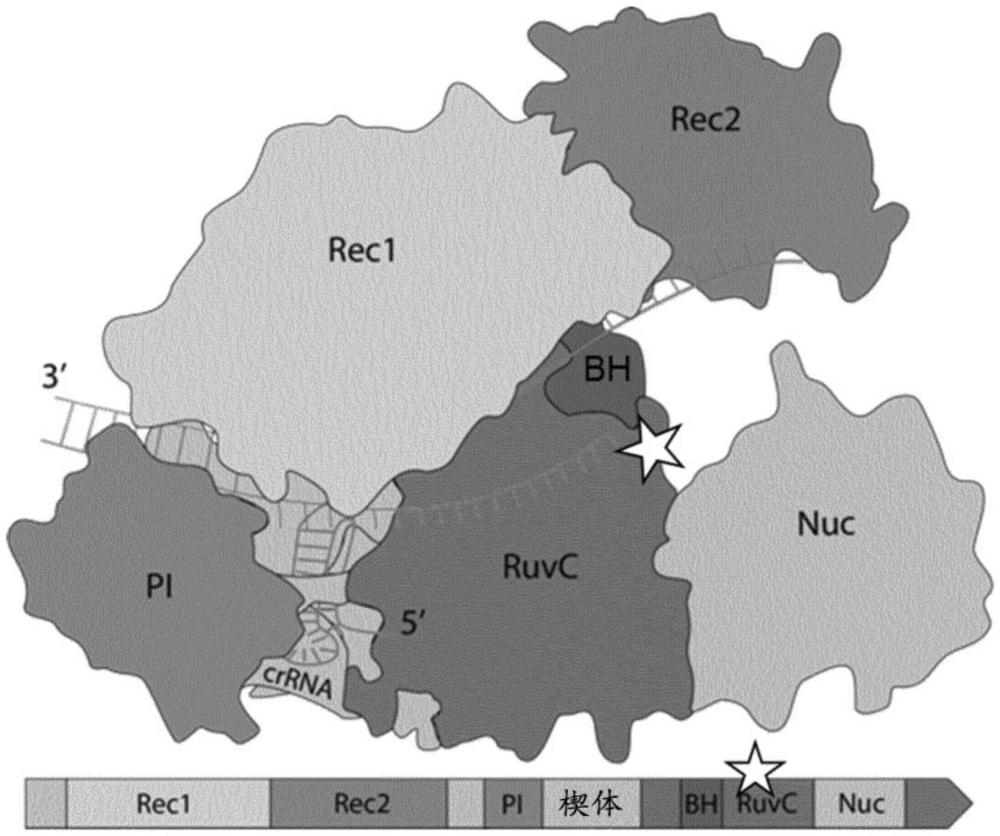
本发明涉及基因基因组编辑领域。特别地,本发明涉及提供具有切口酶(nickase)活性的cas12a酶,以及用于用具有切口酶活性的cas12a酶修饰目的基因组基因座的方式和方法,及其用途。
背景技术:
1、在过去几年中,在dna中产生单链切口而不是双链断裂(dsb)的crispr核酸酶的变体已经作为用于在细胞和生物中靶向基因编辑的通用工具出现。靶标特异性切口产生主要通过cas9切口酶突变体d10a和h840a实现(jinek等人,2012;gasiunas等人,2012)。cas9d10a切割grna靶向链,而cas9 h840a切割非靶向链(jinek等人,2012;gasiunas等人,2012;cong等人,2013;mali等人,2013)。
2、由于切口主要经由高保真碱基切除修复途径修复(dianov和hübscher,2013),因此切口酶能够实现高度特异性编辑。crispr核酸酶通常触发出乎意料的切割,随后在与靶位点具有序列同源性的基因组位点处形成插入缺失(indel)。可以引入成对的切口酶(其通过在相对的dna链上附近产生两个单链断裂而有效地产生dsb)以降低这种脱靶活性。在这种双重切口酶方法中,在每个切割末端上产生长突出端而不是平端。这提供了增强的对精确基因整合和插入的控制。因为两种切口酶(nicking enzyme)必定有效地使其靶dna产生切口,所以与双链切割cas系统相比,成对的切口酶具有显著更低的脱靶效应(ran等人,2013;kuscu等人,2014)。
3、除了减少脱靶编辑之外,还可以利用切口酶来提高精确基因编辑方法(如同源定向修复(hdr)和碱基编辑)的效率。由双链dna切割引发的hdr通常伴随有中靶和脱靶位点处的不需要的插入和缺失(插入缺失)(kosicki等人,2018;shin等人,2017;tsai等人,2015;zhang等人,2015)。切口酶提供了在不刺激nhej的情况下诱导高保真hdr的有吸引力的方法。碱基编辑在不同时形成插入缺失的情况下类似地允许靶位点处的碱基取代。由于碱基编辑器通常不产生dsb,因此它们使dsb相关副产物的产生最小化(komor等人,2016;gaudelli等人,2017)。dna碱基编辑器(be)包括无催化活性的cas核酸酶或切口酶与碱基修饰酶之间的融合物,该碱基修饰酶对单链dna(ssdna)而不是双链dna(dsdna)起作用。在与dna中其靶基因座结合后,指导rna与靶dna链之间的碱基配对导致所称的“r环”中单链dna的小区段的置换(nishimasu等人,2014)。
4、通过脱氨酶对此单链dna泡内的dna碱基进行修饰。为了提高编辑效率,已经设计了许多碱基编辑器以在未经编辑的dna链中引入切口,从而诱导细胞使用经编辑的链作为模板来修复未经编辑的链(komor等人,2016;nishida等人,2016;gaudelli等人,2017)。
5、重要的是,切口酶(如果适当地适配)也可以在最近开发的先导编辑(primeediting)技术中发挥重要作用。先导编辑是一种“搜索和替换”基因组编辑工具,其在不需要dsb或供体模板的情况下介导靶向插入、缺失、所有12种可能的碱基到碱基转化及其组合(anzalone等人,2019)。先导编辑器使用与rna可编程切口酶融合的逆转录酶和先导编辑延伸的指导rna,以将来自pegrna上的延伸的遗传信息直接拷贝到靶基因组基因座中。在这种方法中,使用cas9 h840a切口酶以使非靶链产生切口以暴露3’-羟基基团,该3’-羟基基团引发pegrna上的编辑编码延伸的逆转录直接进入靶位点中。此外,与碱基编辑器非常类似,第三代先导编辑器还使未经编辑的链产生切口以诱导其替换并且进一步提高编辑效率(anzalone等人,2019)。如技术人员所充分了解的,可以根据所希望的靶细胞或构建体来设计和优化pegrna。例如,植物中的先导编辑描述于sretenovic和qi 2021中,并且单子叶植物中的经优化的先导编辑描述于jin等人,2022中。
6、当然,通用碱基和先导编辑器的搜索需要切口酶本身良好的基本功能性(高特异性、宽pam靶向范围、稳定性、低脱靶和高中靶活性)以及切口酶结构域与其他结构域和效应子结构域之间的间隔子的适当空间整合等,使得可以实现在所选的基因组中的靶位点上/处的适当模块化架构和高效活性。
7、目前,crispr-cas系统被分类为两类(类别1和类别2),这两类被细分为六种类型(i型至vi型)。类别1(i型、iii型和iv型)系统在其crispr核糖核蛋白效应子核酸酶中使用多种cas蛋白,并且类别2系统(ii型、v型和vi型)使用单一cas蛋白(nishimasu等人,2017)。除了crispr cas9系统之外,crispr cas12a(或cpf1)系统也已经作为用于大量基因组编辑应用的强大生物技术工具出现。
8、cas9通过两个保守核酸酶结构域ruvc和hnh的组合活性同时切割两条dna链来产生平端dsb(jinek等人,2012;gasiunas等人,2012)。可以通过这些结构域内的关键催化残基的丙氨酸取代来产生cas9切口酶变体:ruvc突变体d10a在靶向链上产生切口,而hnh突变体h840a在非靶向链dna上产生切口(来自酿脓链球菌(streptococcus pygenes)的cas9(spcas9)的氨基酸编号;jinek等人,2012;gasiunas等人,2012;cong等人,2013;mali等人,2013)。
9、最近,对于植物细胞(wo 2021122080a1)已经描述了引入成对的切口强烈地提高了同源定向修复的效率,使得能够通过减少随机插入和/或缺失(插入缺失)将供体dna序列精确引入植物基因组中。此类基于切口酶的方法可以大大减少筛选努力。
10、改善dna的特异性靶向修饰的另一种方法是与供体核苷酸共价连接从而增强hdr效率的指导rna(wo 2017186550a1)。当将供体序列引入靶基因组中时,此类融合核酸分子可以与有效的cas12a切口酶组合以实现最佳效率和特异性。
11、与使用cas9切口酶的早期发现相比,迄今为止cas12a尚未实现(特别是在相关作物植物中未实现)靶标特异性切口产生,并且因此非常需要建立合适的基于cas12a的切口酶工具。
12、与cas9不同,cas12a使用位于ruvc结构域中的单个催化位点依次切割两条dna链,而nuc结构域在底物dna配位中起作用(swarts等人,2017,2019)。与cas9相比,结构组织的这种差异妨碍cas12a的真正切口酶的设计,cas9 crispr核酸酶具有包含两个单独的活性结构域hnh和ruvc(分别催化靶链和非靶链的切割)的两个不同的结构域。
13、在lbcas12a结构中,ruvc活性位点由保守的酸性残基asp832、glu925、asp1180和arg1138形成(yamano等人,2017)。体外切割测定显示d832a、e925a和d1180a突变完全消除了lbcas12a的dna切割活性,而据报道r1138a突变体在体外充当至少部分活性的切口酶,就像r1226a ascas12a的情况(zetsche等人,2015;yamano等人,2016)。还如yamano等人,2017中报道的,lbcas12a和ascas12a在结构和功能上相关。特别地,这些cas12a变体都共享总体结构域架构。另一种报道的切口酶变体包括fncas12a k1013g/r1014g双重突变体,据报道其仅切割靶链(wo 2019/233990)。
14、然而迄今为止,没有证据显示cas12a切口产生变体在体内的特异性切口酶活性,并且因此,没有在多种真核细胞中具有体内高且特异性的切口产生活性的普遍适用的cas12a切口酶。
15、考虑到切口酶在多种基因组编辑工具(hdr、碱基编辑、先导编辑)中的中心作用,开发在体内(包括在植物中)展现出有效dna切口产生的cas12a变体是利用cas12a用于作物遗传改善、治疗应用以及食品和营养科学中的应用的全部潜力的关键。
16、虽然crispr-cas应用在小麦(世界范围最重要的但是难以进行遗传修饰的作物植物之一)中非常困难,但是最近已经开发了用于将供体dna序列精确引入小麦基因组中的有效方法(wo 2021122081a1)。因此,有效且特异性的cas12a切口酶也可能具有改善小麦中的精确遗传修饰的巨大潜力。
17、因此,总体目标是通过合理设计方法并且经由定向进化方法来工程化和鉴定一种或多种cas12a切口酶变体,所述切口酶允许在广泛的原核生物以及还有真核生物的染色体dna中在体外并且特别是还有在体内产生切口(或切口对),其中cas12a切口酶应当具有高特异性切口酶活性和低脱靶活性以及高灵活性以在包括碱基编辑、先导编辑和成对切口酶测定的各种基因组修饰环境中使用,以及总体稳健性和稳定性以提供广泛适用的基因组切口产生工具。
18、定义
19、如本文所用,广谱切口酶活性是指在体外和体内均有效地产生特异性单链dna断裂(切口)的能力,并且具有极少或几乎没有残留核酸酶活性,优选地,其中体外和/或体内(优选体外和体内)残留核酸酶活性小于总酶活性的大约20%、更优选小于大约15%、甚至更优选小于大约10%、并且最优选小于大约5%,其中该总酶活性是具有切口酶活性的给定cas12a酶或其催化活性片段的切口酶活性和核酸酶活性的总和,其中使用合适且合理的反应条件在合适的细胞系统和/或体外系统并进一步在所述细胞系统和/或体外系统的合理限度内在相同的条件下使用相同的一个或多个靶位点通过相同的检测系统和/或方法对具有切口酶活性的给定cas12a酶或其催化活性片段的切口酶活性和核酸酶活性进行确定与比较。技术人员充分了解用于确定cas12a酶的切口酶和核酸酶活性的各种不同的合适方法,包括本文披露的方法。如本文所用,术语“核酸酶活性”是指内切核苷酸活性,其中一种核酸酶效应子能够产生双链断裂,而对于切口酶(为了实现双链断裂)需要两个单独的切口(通过相同的切口酶,或通过至少两种不同的切口酶)。如本文所用,靶链(ts)切口酶活性是指如上所述的切口酶活性,其中至少90%的切口产生发生在靶链中。如本文所用,非靶链(nts)切口酶活性是指如上所述的切口酶活性,其中至少90%的切口产生发生在非靶链中。
20、如本文所用,靶位点是指双链dna的两条链,即与指导rna退火的靶链和互补非靶链,其中该靶位点是针对指导rna的dna段(stretch),该指导rna与该靶链具有合适的互补性,其中,在其中至少两种相容的指导rna被设计为允许一种或至少两种cas酶的协调作用的实施例中,该靶位点是指该至少两个dna段,对于其中的每一个,一个指导rna与靶链具有互补性,并且该靶位点进一步包括在所述至少两个dna段之间的任何dna序列(还参见图7a),其中所述至少两个dna段(一个指导rna与其中的每一个具有互补性)也可以重叠或者可以是相同的。
21、如本文所用,“在靶位点处或附近”是指在靶位点内或在靶位点旁边至多10bp、至多20bp、至多30bp或至多40bp的dna部分,包括两个方向。
22、“供体修复模板”或“供体模板”、或“供体dna”或仅“供体”是指可以被提供来允许和介导hdr的核酸模板,其可以用于实现靶基因座的无错误修饰和/或引入外来核酸序列(如转基因)。该至少一种供体修复模板可以包含或编码双链核酸序列和/或单链核酸序列。该至少一种供体修复模板可以包含或编码rna和/或dna序列。该至少一种供体修复模板可以包含或编码对称或不对称同源臂。在某些实施例中,该至少一种供体修复模板可以进一步包含至少一种经化学修饰的碱基和/或骨架,如荧光标记物和/或经硫代磷酸酯修饰的骨架。用于各种目的的供体修复模板的设计和使用是技术人员熟知的。
23、如本文所用,术语“疾病状态相关靶位点”是指某些等位基因、变体或突变实际上或潜在地引起、影响至少一种身体和/或精神疾病、小病、障碍或不良病症或倾向或其进展或预后或者可能是至少一种身体和/或精神疾病、小病、障碍或不良病症或倾向或其进展或预后的风险因素的任何靶位点。疾病状态相关靶位点可以例如是在蛋白质编码基因内包含错义或无义突变的靶位点,或者它可以是包含多态性(如单核苷酸多态性)的变体的靶位点,这种关联可能是患上某种疾病的风险因素。
24、术语“指导rna”可以是指包含cas蛋白结合区和靶向区的任何rna,并且只要靶核苷酸序列位于适合于相应cas蛋白的pam序列的旁边,该指导rna就能够将cas蛋白引导至与指导rna的靶向区充分互补的靶核苷酸序列。对于cas12a系统,术语“指导rna”、“crrna”、“grna”或“sgrna”可互换使用。对于在如本领域已知的在天然环境中使用双分子指导rna(如crrna和tracrrna)的系统和/或方法,术语指导rna是指两种rna分子。一旦描述了包括cas酶和同源指导rna(crrna或crrna::tracrrna)的crispr效应子系统,技术人员因此了解哪种类型的指导rna用于哪种类型的cas酶,例如cas12a系统使用单一crrna,而cas12e系统类似于cas9系统使用crrna::tracrrna双链体,然而其中crrna::tracrrna双链体可以被合成的单一指导rna分子模拟。此外,技术人员充分了解出于所需目的设计、表达/合成和调整指导rna。特别地,如本文提供的(n)cas12a酶及其(n)cas12直向同源物的突变将不会影响用于给定ncas12a酶或ncas12直向同源物的同源指导rna的总体设计和相互作用模式。在涉及先导编辑器或先导编辑器复合物的实施例中,指导rna可以是pegrna(先导编辑指导rna),并且可以进一步包含引物结合位点(pbs)和/或逆转录酶模板序列。适合于各种不同cas系统的指导rna(包括pegrna)的设计是技术人员熟知的。
25、当关于比较两个或更多个核酸或氨基酸分子使用时,“同一性”意指所述分子的序列具有一定程度的序列相似性,这些序列是部分相同的。
26、酶变体可以通过与亲本酶比较时它们的序列同一性进行定义。序列同一性通常以“序列同一性%”或“同一性%”的形式提供。为了在第一步中确定两个氨基酸序列之间的同一性百分比,在这两个序列之间生成成对序列比对,其中这两个序列在它们的完整长度上比对(即,成对全局比对)。用实施needleman和wunsch算法(j.mol.biol.[分子生物学杂志](1979)48,第443-453页)的程序,优选地通过使用具有程序默认参数(空位开放=10.0,空位延伸=0.5和矩阵=eblosum62)的程序“needle”(欧洲分子生物学开放软件套件(european molecular biology open software suite,emboss))生成比对。用于本发明目的的优选比对是可以从中确定最高序列同一性的比对。
27、以下实例意在说明两个核苷酸序列,但相同的计算适用于蛋白质序列:
28、序列a:aagatactg,长度:9个碱基
29、序列b:gatctga,长度:7个碱基
30、因此,较短的序列是序列b。
31、产生显示完整长度的两个序列的成对全局比对,结果是
32、序列a:aagatactg-
33、||||||
34、序列b:--gat-ctga
35、比对中的“i”符号表示相同的残基(这意指dna的碱基或蛋白质的氨基酸)。相同残基的数目为6。
36、比对中的“-”符号表示空位。序列b内通过比对引入的空位数目为1。序列b边界处通过比对引入的空位数目为2,而序列a边界处引入的空位数目为1。
37、显示完整长度的比对序列的比对长度为10。
38、因此,根据本发明,产生显示完整长度的较短序列的成对比对,结果是:
39、序列a:gatactg-
40、||||||
41、序列b:gat-ctga
42、因此,根据本发明,产生显示完整长度的序列a的成对比对,结果是:
43、序列a:aagatactg
44、||||||
45、序列b:--gat-ctg
46、因此,根据本发明,产生显示完整长度的序列b的成对比对,结果是:
47、序列a:gatactg-
48、||||||
49、序列b:gat-ctga
50、显示完整长度的较短序列的比对长度为8(存在一个空位,该空位被计入该较短序列的比对长度)。
51、因此,显示完整长度的序列a的比对长度为9(意味着序列a是本发明的序列)。
52、因此,显示完整长度的序列b的比对长度为8(意味着序列b是本发明的序列)。
53、比对两个序列后,在第二步中,根据产生的比对确定同一性值。为了此描述的目的,同一性百分比通过以下计算:同一性%=(相同残基/显示完整长度的本发明的相应序列的比对区域的长度)*100。因此,与根据本实施例的两个氨基酸序列的比较相关的序列同一性是通过将相同残基的数目除以显示完整长度的本发明的相应序列的比对区域的长度来计算的。该值乘以100得到“同一性%”。根据上文提供的实例,同一性%:对于seq a是本发明的序列,为(6/9)*100=66.7%;对于seq b是本发明的序列,为(6/8)*100=75%。
54、“插入缺失(indel)”是与nhej修复dsb相关的在生物基因组中碱基的随机插入或缺失的术语。它被归类为属于小的遗传变异,测量的长度为1至10 000个碱基对。如本文所用,它是指在靶位点中或在靶位点附近(例如,在靶位点上游和/或下游小于1000bp、900bp、800bp、700bp、600bp、500bp、400bp、300bp、250bp、200bp、150bp、100bp、50bp、40bp、30bp、25bp、20bp、15bp、10bp或5bp处)随机插入或缺失碱基。
55、如本文所用,术语体外是指不在活细胞内部,优选在无细胞系统中进行的方法或应用或程序的状态或特质。体外方法、应用或程序典型地用生物材料(如已经从细胞中纯化和/或人工加工或合成的核酸、多肽等)通常在包含合适的缓冲液系统和合适的反应组分的反应管或反应室中进行。
56、如本文所用,术语体内是指包括操纵至少一种活细胞(包括在细胞培养中生长的细胞)(如将crispr组分引入活细胞中和所述细胞内潜在的基因组切口产生、双链切割和/或修饰)的方法、应用或程序的状态或特质。体内方法、应用或程序可以随后是例如细胞裂解后纯化的dna的体外分析。因此,如本文所用,体内并不一定意味着方法在活生物内进行,该体内方法可以在体外环境(如体外细胞培养)中进行。
57、如本文所用,术语离体是指针对从生物提取的活细胞和/或活组织的方法、应用或程序的状态或特质,其中所述活细胞和/或活组织可以在离体方法、应用或程序之后重新插入从中提取所述活细胞和/或活组织的生物中。
58、如本文所用,术语“偏移”是指被设计为允许一种或至少两种cas酶的协调作用的两个指导rna的结合位点之间的碱基对的数目(参见图7a,显示+5bp的示例性偏移)。
技术实现思路
本文地址:https://www.jishuxx.com/zhuanli/20241021/317709.html
版权声明:本文内容由互联网用户自发贡献,该文观点仅代表作者本人。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如发现本站有涉嫌抄袭侵权/违法违规的内容, 请发送邮件至 YYfuon@163.com 举报,一经查实,本站将立刻删除。